自动生成吸附模型
本文参考文献: A high-throughput framework for determining adsorption energies on solid surfaces
https://www.nature.com/articles/s41524-017-0017-z
Gerbrand Ceder, Kristin A. Persson等人领导团队多年来一直致力于高通量计算方法,和materials project数据库的发展,https://materialsproject.org。
2017年Kristin A. Persson 和 Joseph H. Montoya发展了表面吸附模型的高通量计算模块集成到pymatgen程序包里,使得表面吸附分子的数据库构建成为可能。这篇文章就介绍一下用pymatgen实现表面高通量计算的方法。
其中有几个关键点:
- 通过API自动得到数据库里的晶胞结构。(以后会另外写一篇文章专门介绍Materials project的API)
- 自动切割表面,要考虑的问题非常多:比如,表面的厚度,最表层暴露的原子,晶面指数,真空层厚度,slab模型的位置等等。
- 自动识别不同的吸附位点,比如:top,bridge,hollow。识别要基于表面的对称性,这就需要程序有可靠的对称性识别算法,对于slab模型的整体对称性和不等价吸附位点能准确判断。
- 自动添加吸附原子/分子,并保存好POSCAR文件。
- 批量提交任务。
手动建立一个Ni晶胞,切(111)面,并自动识别不等价的吸附位点。
from pymatgen import Structure, Lattice, MPRester, Molecule from pymatgen.analysis.adsorption import * from pymatgen.core.surface import generate_all_slabs from pymatgen.symmetry.analyzer import SpacegroupAnalyzer from matplotlib import pyplot as plt from pymatgen.ext.matproj import MPRester from pymatgen.io.vasp.inputs import Poscar #Structure.from_spacegroup建立一个类,通过空间群和坐标参数生成一个Structure对象。 fcc_ni = Structure.from_spacegroup("Fm-3m", Lattice.cubic(3.5), ["Ni", "Ni"], [[0, 0, 0], [0.5, 0.5, 0.5]]) #然后对这个晶胞结构进行自动切面,max_index=1得到最大的不等价表面,比如(1, 1, 1) (1, 1, 0) (1, 0, 0),min_slab_size=8.0, min_vacuum_size=15.0控制晶胞的厚度和真空层。 slabs = generate_all_slabs(fcc_ni, max_index=1, min_slab_size=8.0, min_vacuum_size=15.0) ni_111 = [slab for slab in slabs if slab.miller_index==(1,1,1)][0] #然后用AdsorbateSiteFinder找到有可能的吸附位点。把print(ads_sites)前的注释拿掉,就能打印出所有的吸附位置。 asf_ni_111 = AdsorbateSiteFinder(ni_111) ads_sites = asf_ni_111.find_adsorption_sites() # print(ads_sites) assert len(ads_sites) == 4 #用matplotlib就能直接画出所有的可能吸附位点了: fig = plt.figure() ax = fig.add_subplot(111) plot_slab(ni_111, ax, adsorption_sites=True) plt.savefig('111.png', img_format='png')添加吸附分子或原子:
想要吸附原子或者分子,点定义一个变量:
做HER反应吸附H原子
adsorbate = Molecule("H", [[0, 0, 0]])做ORR,OER反应吸附O,OH,OOH
OH = Molecule("OH", [[0, 0, 0], [-0.793, 0.384, 0.422]]) O = Molecule("O", [[0, 0, 0]]) OOH = Molecule("OOH", [[0, 0, 0], [-1.067, -0.403, 0.796], [-0.696, -0.272, 1.706]])用generate_adsorption_structures函数产生吸附结构,用matplotlib画图:
fig = plt.figure() ax = fig.add_subplot(111) adsorbate = Molecule("H", [[0, 0, 0]]) ads_structs = asf_ni_111.generate_adsorption_structures(adsorbate, repeat=[1, 1, 1]) plot_slab(ads_structs[0], ax, adsorption_sites=False, decay=0.09) plt.savefig('111-H.png', img_format='png')使用API,从Materials project得到晶体结构:
# Import statements from pymatgen import Structure, Lattice, MPRester, Molecule from pymatgen.analysis.adsorption import * from pymatgen.core.surface import generate_all_slabs from pymatgen.symmetry.analyzer import SpacegroupAnalyzer from matplotlib import pyplot as plt from pymatgen.ext.matproj import MPRester from pymatgen.io.vasp.inputs import Poscar # Note that you must provide your own API Key, which can # be accessed via the Dashboard at materialsproject.org mpr = MPRester('yourPrivateKey') fig = plt.figure() axes = [fig.add_subplot(2, 3, i) for i in range(1, 7)] mats = {"mp-23":(1, 0, 0), # FCC Ni "mp-2":(1, 1, 0), # FCC Au "mp-13":(1, 1, 0), # BCC Fe "mp-33":(0, 0, 1), # HCP Ru "mp-5229":(1, 0, 0), # Cubic SrTiO3 "mp-2133":(0, 0, 1)} # Wurtzite ZnO for n, (mp_id, m_index) in enumerate(mats.items()): struct = mpr.get_structure_by_material_id(mp_id) struct = SpacegroupAnalyzer(struct).get_conventional_standard_structure() slabs = generate_all_slabs(struct, 1, 5.0, 2.0, center_slab=True) slab_dict = {slab.miller_index:slab for slab in slabs} asf = AdsorbateSiteFinder.from_bulk_and_miller(struct, m_index) plot_slab(asf.slab, axes[n]) ads_sites = asf.find_adsorption_sites() sop = get_rot(asf.slab) ads_sites = [sop.operate(ads_site)[:2].tolist() for ads_site in ads_sites] axes[n].plot(*zip(*ads_sites), color=’k’, marker=’x’,markersize=10, mew=1,\ linestyle=’’, zorder=10000) mi_string = "".join([str(i) for i in m_index]) axes[n].set_title("{}({})".format(struct.composition.reduced_formula, mi_string)) axes[n].set_xticks([]) axes[n].set_yticks([])想要导出POSCAR文件用open().write()即可,这是我批量到处POSCAR的方式:
open('POSCAR' + mp_id + '-' + "{}({})".format(struct.composition.reduced_formula, mi_string) + '-' + str(count), 'w').write(str(Poscar(reorient_z(slabmodel))))
实战演练,金属和合金表面HER反应高通量计算:
笔者读了Kristin A. Persson文章之后一直在思考一个问题,既然这个功能2017年已经开发完善了,但是这都2019年了,迟迟不见表面吸附数据库出现。经过笔者实战尝试时候终于懂了为什么。如果只考虑Ni和Pt两种元素,他们之间可能形成 Ni,Pt,NiPt,Ni3Pt,NiPt3这5中组成状况,每种晶体就算只考虑Miller最高到(1, 1, 1),这样每种晶体也有三种表面,然后每种表面有4到10几种不同的吸附位点,这样算下来完成Ni/Pt二元合金的HER计算,就生成了5 3 10 = 150个结构,如果考虑到整个周期表中的金属和二元合金的话,组合情况至少要多100倍,这样就是上万个结构。每个结构如果要在20核的单节点上算5小时,算下来就是上百万的核时。壕如 Gerbrand Ceder, Kristin A. Persson也不敢轻易的去尝试表面数据库的搭建,所以笔者也只是满足于自动生成了上千种POSCAR文件结构就浅尝辄止了。
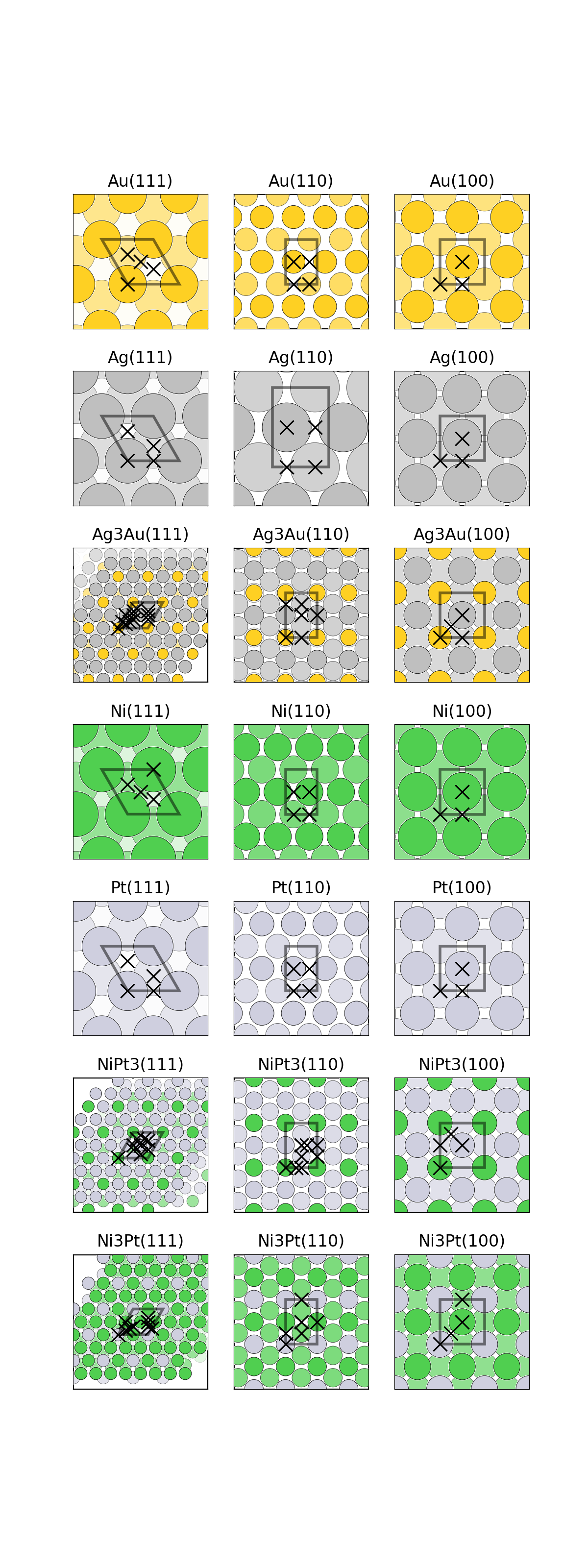
如果真要做高通量,自己去手动输入mp-23 mp-5229这样的编号显然是比较笨的方法,用元素组成,然后自动去数据库里下载结构文件时更优方法:
struct = mpr.get_structures('Ni-Pt-', final=True)
到这里,我们已经把高通量计算最难的结构批量搭建完成了,后面就是自动生成POTCAR,KPOINTS,INCAR文件了,这些东西用VASPKIT或者pymatgen,或者自己写个小循环就能轻易搞定的了。流程如下:
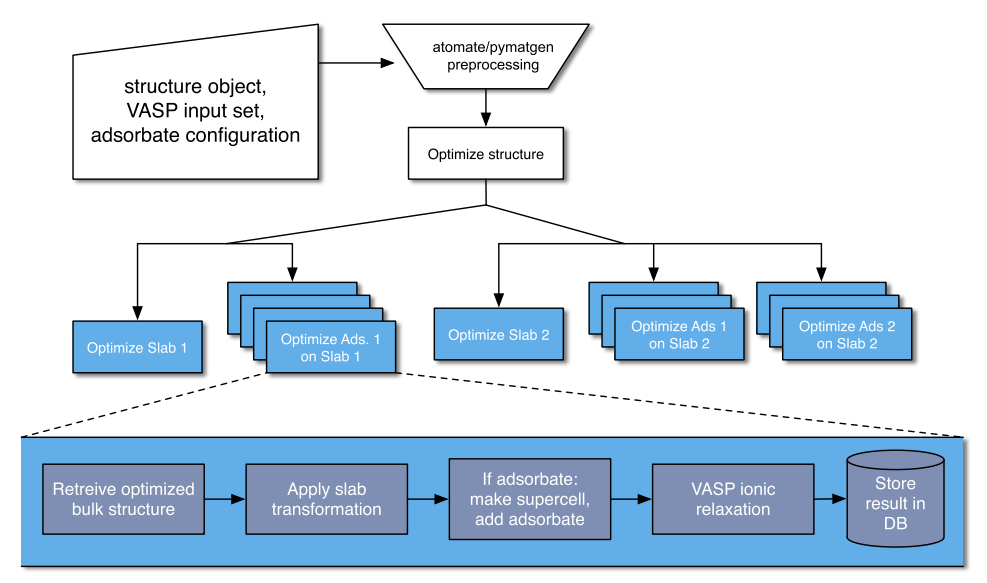
VASP批量计算完成以后,还有重要的内容就是后处理了,首先就是要把后处理程序化,这一点pymatgen和VASPKIT都做得非常好,把这些数据批处理以后再导入到materials project数据库里或者自己搭建一个新得表面模型数据库。然后,怎么在浩如烟海的计算结果里日提取出规律性的东西,这就是另外一回事了。