频率计算
虚频也作为收敛的一个判据
应用场景:
- 确定结构是否稳定;
- 看振动方式和大小,用来和实验对比,棋博士最新的文章就是一个非常好的例子;
- 反应热,反应能垒, 吸附能等的零点能(ZPE)矫正;
- 确认过渡态(有一个振动的虚频)
- 热力学中计算entropy,用于计算化学势,微观动力学中的指前因子和反应能垒
计算步骤:
依照前面所述进行优化
将CONTCAR复制为POSCAR
修改INCAR,主要修改参数如下:
IBRION=5 #freq calc POTIM=0.02 #一个更小的值,default:0.015 NSW=1 # any value which >1 NFREE=2 # do not set NFREE=1,添加这一个参数,表明原子在某一方向上正反两个方向移动 # NCORE=4 并行计算频率时VASP会罢工 EDIFFG=1E-6 #严格一些,需要保证准确查看频率分析的OUTCAR,OSZICAR等输出文件
当你设置了NFREE=2的时候,频率计算需要1+N*6步。N为体系中的振动的原子数
vi command:: set nu 显示文本的行数,取消行数可以通过 :set nonu
固定催化剂部分计算振动频率:
三个方向都允许:T T T 三个方向上都不允许为 F F F x移动,y和z方向上固定为: T F F x和y方向上固定,z方向振动: F F T
通过Vim的实现 :
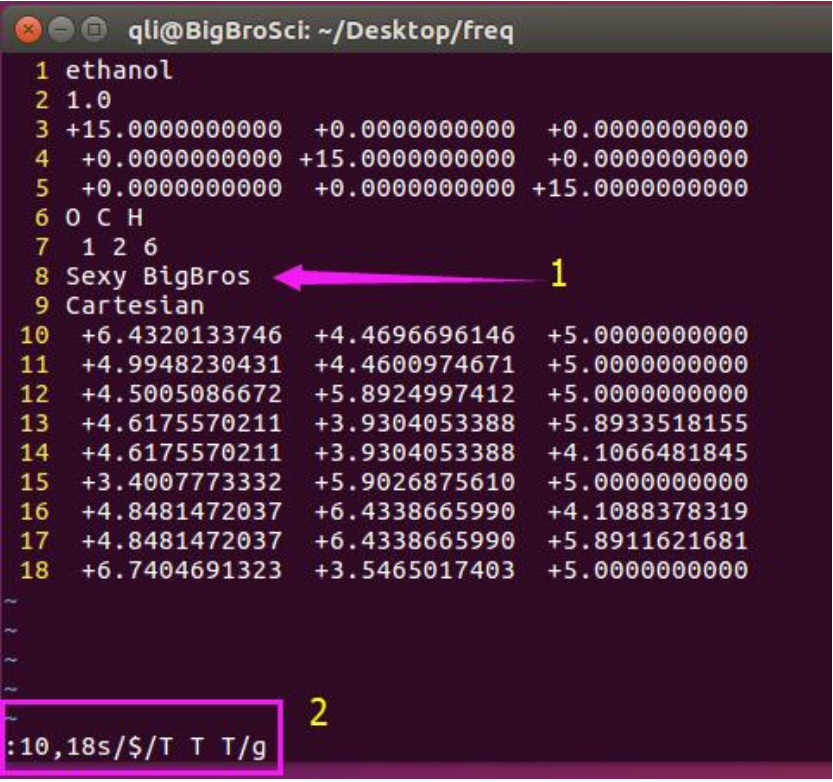
图中1:插入一行,告诉VASP我们要选择性的固定某些原子或者在某些方向上; 图中2: 10,18s 中 s 代表替换(substitute)的意思,这里表示我们选中了第10到18行,10和18之间有个逗号表示连续;10,18s后面用一个 / 分开,紧跟着你我们要替 换的内容;$在这里是末尾的意思,
$/T T T我们要把每一行的最后替换成 T T T 后面再用一个/分开,加上g 表示 global 全部替换的意思。
p4vasp亦可
振动频率可视化:
Linux安装jmol
jmol.sh OUTCAR工具-原子库选择器
也可以右键-模型中选中
原子库中选择最下方:振动,开启振动
OUTCAR中的原信息:
1)1 f = 代表第一个振动模式,细心的你仔细观察,会发现每一行有四个单位的数值:THz, 2PiTHz, cm-1,和meV,这四个是完全等同的; 2)下面一行为坐标XY Z和每个原子在x y z方向上的振动大小; 3)X Y Z下面的数字为结构的坐标信息(Cartesian坐标系),dx dy dz 为振动的具体数值; 4)后面的振动模式的频率和第一个的格式一样。
频率单位的换算 :
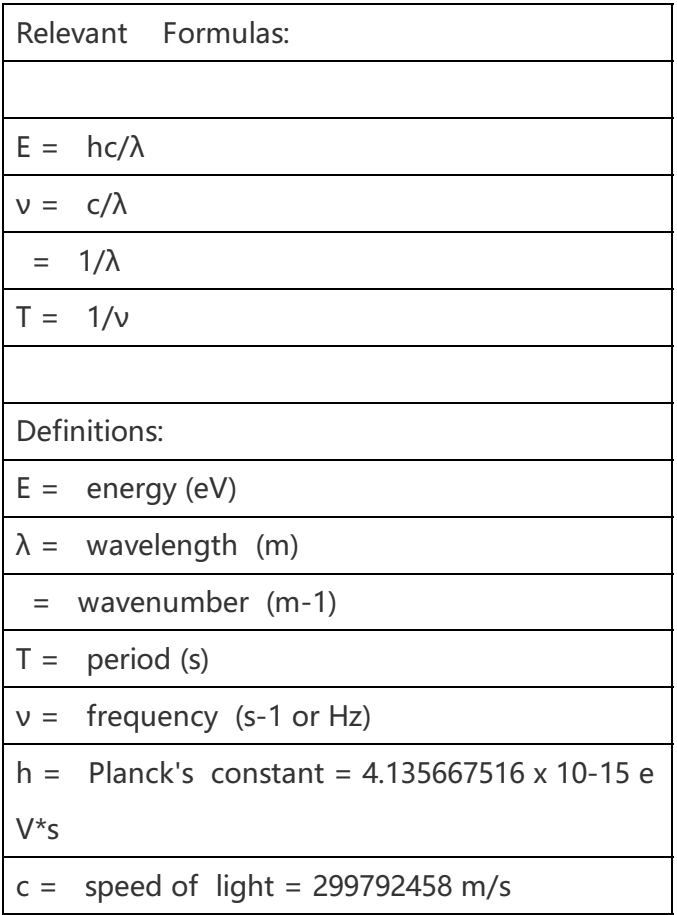
OUTCAR频率信息的提取 :
grep THz OUTCAR grep 2PiTHz OUTCAR grep cm-1 OUTCAR grep meV OUTCAR综合这些因素,对于分子的振动频率来说(注意:声子谱不适用)一般低于100cm-1的频率可以忽略。严格点可以降到 50 cm ,也就是说:如果你在计算中发现有个50cm 左右的虚频,完全可以不考虑
零点能校正 :
输出共有11列(列之间用空格分开):我们要的零点能在第10列,使用下面的命令:
grep 'f =' OUTCAR | awk '{print $10}如果想同时输出第1和10两列:
grep 'f =' OUTCAR | awk'{print $1 " "$10}'1 和 $10 之间有2个 双引号:" ",两个双引号里面有一个空格用来分开),否则两列会连在一起。
这里我们提取的能量为:hv !!!而零点能为1/2 hv!!!
将所有振动的能量求和:
grep 'f =' OUTCAR | awk '{print $10}' | paste -sd+ |bc不要忘记除以2,换算为eV除以1000
A)首先结构优化完毕后我们会得到分子的能量:E0 B)频率计算后我们会得到分子的零点能:ZPE C)零点能校正之后的乙醇分子能量为: E_ZPE = E0 + ZPE D)A和B得出的结果直接相加即可,不要想太多。
怎么计算过渡态和反应热的零点能校正
对一个反应:IS --> TS --> FS IS: Initial State 反应物 TS: Transition State 过渡态 FS: Final State 产物
1)优化反应物IS和产物FS的结构,获得能量:E(IS),E(FS); 2)对反应物和产物进行频率计算,获得各自的零点能:ZPE(IS), ZPE(FS)。 3)搜索过渡态,获得结构和能量E(TS); 4)过渡态频率分析,获得零点能ZPE(TS)
不考虑零点能的反应能垒(E )和反应热(ΔE): E = E(TS) – E(IS) ΔE =E(FS) – E(IS)
考虑零点能校正: $E = E{ZPE}(TS) – E{ZPE}(IS) = E(TS) +ZPE(TS) – E(IS) – ZPE(IS) = E + ZPE(TS) – ZPE(IS)$同理: ΔE =ΔE + ZPE(FS) –ZPE(IS)
两个处理方式: a. 先获取未校正的结果,然后把零点能各自相减; b. 先将各个物种进行零点能校正,然后在计算反应能垒或者反应热效果是一样的
频率计算的时候,是不是体系中所有的原子都放开 :
例子A: 乙醇在Cu(111)表面上的吸附,计算吸附热的零点能校正 $\ce{CH3CH2OH + Cu(111) -> Cu(111)-CH2CH3OH}$ 此时,Cu(111)表面我们在计算频率的时候是要固定住的!只振动乙醇分子即可。 例子B: 计算CO在Cu(111)表面上的吸附: 同A,固定Cu(111) 表面,如果你只关心CO在垂直表面上的振动,那么CO的xy方向便可以固定住,在坐标后面为:F F T 例子C: 苯酚在Cu(111)表面上O—H键断裂活化能的零点能校正: 在这里我们拿苯酚作为例子,很多时候,计算的对象比较大,全部频率优化非常耗时,那我们就得选择性地固定住一部分,只关心关键的局域部分。这个例子中我们主要讨论零点能对O—H键断裂活化能的影响,因此我们可以把苯环的部分固定住,只放开O和H原子进行振动。
还有一点要注意的是:IS,TS和FS中,所固定和放开的原子必须一致!!!
获取虚频命令:
grep 'f/i' */OUTCAR | awk '{print $1 "\t " $2 "\t" $8 "\t " $9 "\t" $10 "\t" $11}'获取时间命令:grep Elapsed */OUTCAR | sort -n获取零点能:for i in * ; do echo $i $(cd $i ; fsum ; cd $OLDPWD);done | sort –nPOTIM太小的时候,对零点能影响很大
POTIM 太大的时候,会搞出来超大号的虚频
5.1 IBRION = 5 (告诉VASP我们要算频率) 5.2 POTIM = 0.015 5.3 NFREE = 2 5.4 ENCUT和原来一样 5.5 PREC = Normal 5.6 EDIFF = 1E-5 或者 1E-6 5.7 KPOINTS Gamma点即可。