表面吸附以及模型的搭建思想
本章搭建过程自动化可参见后面工具分篇
吸附是指某种气体,液体或者被溶解的固体的原子,离子或者分子附着在某表面上。这一过程使得表面上产生由吸附物构成的膜。吸附不同于吸收,吸收是指作为吸附物的液体浸入或者溶解于另一液体或固体中的过程。吸附仅限于固体表面,而吸收同时作用于表面和内部
需要注意的有3个方面 :
熟悉VASP的POSCAR格式: 每一行的内容所代表的意思以及它们之间的相互联系: A) 元素行,原子数目以及原子的坐标之间的关系; B) Selective Dynamic 行与 T T T, F F F 等原子固定的关系; C)Cartesian坐标和Direct坐标的区别等等; D)POSCAR 与 KPOINTS的关系; 熟悉POSCAR或者CONTCAR的格式可以使你在搭建模型过程中游刃有余。
Slab模型在 Z方向上: slab的厚度和真空层的厚度 这两点一方面决定了我们计算量的大小,另一方面,对于不同的体系;我们需要不同厚度的slab模型来保证计算的准确性。例如:对于金属体系来说:开放的表面往往需要更多的层数。所以,在准确性和计算量上,我们要合理地权衡和取舍。可以通过一些基本的测试工作,以及参考他人发表的计算参数来确定。
Slab模型在XY方向上:
A)表面的大小:这个主要影响覆盖度,计算的工作量,
B)表面的吸附位点:比如下图中面心立方金属的111表面,有top,bridge,fcc 和hcp这几个位置,在搭建模型的时候要考虑吸附物种分别在这几个位置上的情况;
C)吸附物种与表面的结合情况:不同的分子构型?用什么原子?哪一部位和表面接触?初始猜测的键长多少? 等等
这些对于你搭建合理的模型至关重要。一个合理的模型可以极大地降低计算工作量,提高你的计算成功率。当然,这需要一个扎实和良好的化学基本功来作为保障。
为了得到合理的吸附距离,可用一个原子和吸附物质使用1个Gamma点建立模型优化(高通量自动建立模型参见Chapter9)
合理构型的获得:固定slab模型中的所有原子!所有原子!所有原子! 放松EDIFF和EDIFFG,表面吸附物质弛豫
正式计算的时候 :
- 结构方面:将CONTCAR 复制成 POSCAR ;前面一步我们还固定了表层的原子,此时我们需要放开。使用sed命令:
sed -i ‘12,13s/F/T/g’ POSCAR; - KPOINTS: 使用前面slab优化时用的
- INCAR中,如果前面一步的计算精度较低,这时,我们就要提高一下了
- POTCAR 不变
- 结构方面:将CONTCAR 复制成 POSCAR ;前面一步我们还固定了表层的原子,此时我们需要放开。使用sed命令:
吸附能的计算 :
从前面的计算中可以获取哪些能量 :
1) Cu(111) slab 的能量 2) slab上有O吸附时,体系优化完的能量 3) $\ce{O2}$分子 4) 以及O原子在气相中的能量
什么是吸附能 :
1) 先说下终态:这里说的终态就是O原子在表面吸附,并且优化完的构型。我们标记为 slab+O,它的能量为 E(slab+O)
2) 初始状态。我们计算O的吸附能,起始状态是$\ce{O2}$和 纯净的slab表面。因为在实际反应中,$\ce{O2}$在气相中解离成2个O原子,然后再吸附,这种可能性微乎其微。 前面我们讲了$\ce{O2}$分子的优化,这里我们直接把$\ce{O2}$分子的能量拿来用。起始的两个结构能量分别标记为:E($\ce{O2}$) 和 E(slab)。 由于在终态的吸附结构上面,我们只有1个氧原子, 所以初始态我们要用O 能量的一半: E($\ce{O2}$)/2
3) 注意:文献报道里面,也有很多人用O原子在气相中的能量来计算O的吸附能。这样做的话,其物理意义为:$\ce{O2}$需要先解离成O原子,然后O原子再吸附。此时,初始状态为分解的O原子,因此$\ce{O2}$分子解离的能量没有考虑在内。由于$\ce{O2}$解离是吸热反应,忽略掉解离能,会导致O的吸附能很强。单个O原子的能量标记为:E(O)
4) 所以,大家在计算的时候,一定要把自己的计算公式标出来,此外,大家看到直接用O原子能量计算出来的吸附能,不要用O 作为参考能量的结果进行对比。
5) 这两个计算方法,哪个对,哪个错呢? 答:都是对的。因为$\ce{O2}$的解离能是一个常数,加上去(用O 能量作为起始)或者忽略掉(用O原子能量作为起始)得到的结果之间的区别无非也是这个常数。 (看相对值)
计算吸附能(单个O原子) : $$ (formula\ 1)\ E_{ads}(O) = E(slab+O) – E(slab) – E(O_2)/2 $$
$$ (formula\ 2)\ E_{ads}(O) = E(slab+O) – E(slab) – E(O) $$
$E(slab+O)$: O 在Slab上,优化完结构的能量; $E(slab)$: 优化的slab的能量; $E(O_2)$: O 分子在气相中的能量; $E(O)$: O原子在气相中的能量;
O 分子优化计算的时候,要注意 :
i) 从数据库获取O 分子的键长,作为初始值; ii)ISPIN = 2 iii)ISMEAR = 0; SIMGA = 0.05 iv) IBRION= 2; POTIM = 0.1
O 原子优化计算的时候,注意的计算细节与前面相同。额外注意的是:
i) O 原子再怎么优化还是一个O原子,所以上一行表述是不恰当的,应该是O原子的单点计算; ii) box的尺寸为:$131415$ Å 或者$13.113.213.3$ Å 但绝对不能为$131313$这种正方体的。
防止拥挤,建议下面用超胞计算(2*2)
扩胞后,很多人问计算吸附能的步骤,不清楚该怎么样进行。 第一步:优化扩胞后的Slab 模型: 因为扩抱前已经relax过了,所以这一步很快就会结束;得到新的slab能量。 第二步:在第一步基础上搭建吸附模型; 然后优化,得到slab+吸附物种的能量。 第三步:套公式,计算吸附能。
当然了,如果你经验丰富,计算的够多了,可以尝试这样做: 第一步:优化扩胞后的Slab 模型: 因为扩抱前已经relax过了,所以这一步很快就会结束;得到新的slab能量。 第二步:在扩胞后的slab模型上直接搭建吸附模型,然后优化,得到slab+吸附物种的能量。 第三步:套公式,计算吸附能
什么是覆盖度? Surface coverage(θ)
从字面上理解,就是表面被吸附物种覆盖的程度。一个固定大小的表面,如果上面吸附的某一物种越多,那么说它的覆盖度也就越大。也可以理解为:该物种在表面上的浓度也就越大。在多相催化的过程中,吸附是第一步。而在催化剂表面上,其活性位点数量是有限的(N个),如果N1个活性位点被吸附物种占据了,那么此时的覆盖度:θ= N1/N。
slab的表面我应该放开还是固定呢?
严格来说,slab的表层原子应该是弛豫的,也就是坐标后对应的是 T T T,这样更符合物理化学意义,因为催化剂表面的原子不可能会不动。厉害的时候,吸附物种还会导致表面重构。所以你在阅读文献,会发现理论部分都会写:top two layers 放开来模拟表面,底部的原子固定来模拟体相等等这样类似的话。 但是,将表面原子放开后,会导致计算量的增加。如果你的计算量很大,可以测试下放开,或者固定前后,吸附能的差别。基本上应该在一个很小的范围之间。这样你的心里就有数了。
注意:在计算纯的slab能量时,表面应该是放开的。不要直接拿刚刚切好的表面,直接固定住就开始算吸附。
sed命令进阶版:
for i in {1..4}; do sed -i "$((8+4*$i)), $((9+4*$i))s/T T T/F F F/g" POSCAR也可以用excel批量固定原子
还可以使用sortcar.py 将坐标按照z方向大小排列。输出文件为XXX_sorted,其中 XXX为我们想要排列的POSCAR或者CONTCAR
#!/usr/bin/env python from collections import defaultdict import numpy as np import sys in_file = sys.argv[1] ### READ input file ### def read_inputcar(in_file): f = open(in_file, 'r') lines = f.readlines() f.close() ele_name = lines[5].strip().split() ele_num = [int(i) for i in lines[6].strip().split()] dict_contcar = {ele_name[i]:ele_num[i] for i in range(0, len(ele_name))} dict_contcar2 = defaultdict(list) for element in ele_name: indice = ele_name.index(element) n_start = sum(ele_num[i] for i in range(0, indice+1)) - dict_contcar.get(element) +1 n_end = sum(ele_num[i] for i in range(0, indice+1)) +1 dict_contcar2[element].append(range(n_start, n_end)) return lines, ele_name, ele_num, dict_contcar2, dict_contcar def get_elements(ele): lines, ele_name, ele_num, dict_contcar2, dict_contar = read_inputcar(in_file) coord_total = [] my_list = [] my_dict = {} for j in dict_contcar2.get(ele)[0]: coord_list = lines[j+8].strip().split()[0:3] tf_list = lines[j+8].strip().split()[3:] my_list.append(coord_list) dict_key = '-'.join(coord_list) my_dict[dict_key] = tf_list data = np.array(my_list) data=data[np.argsort(data[:,2])] for k in data: coord = ' '.join(k) tf = ' '.join(my_dict.get('-'.join(k))) coord_total.append(coord + ' ' + tf ) return coord_total ## Generate the New POSCAR file def Get_and_Save_lines(file_name, start_line, end_line): f = open(file_name) lines = f.readlines() for line in lines[int(start_line):int(end_line)]: file_out.write(line.rstrip()+'\n') out_name = in_file + '_sorted' file_out = open(out_name, 'w') Get_and_Save_lines(in_file, 0, 9) ele_name = read_inputcar(in_file)[1] dict_contcar = read_inputcar(in_file)[-1] for i in ele_name: if dict_contcar.get(i) > 1 : file_out.write('\n'.join(get_elements(i))) else: file_out.write('\n %s \n' %(' '.join(get_elements(i))))前面我们发现将p(1x1)-Cu(111)的表面扩展成p(2x2)后,由于O的覆盖度降低了,O原子的吸附能能+1.2 eV降低到 0.2 eV作用,说明O原子更加容易吸附在表面上了。当你仔细观察p(2x2)的表面,你会发现,表面上不仅仅有Cu原子的上方可以放O原子,还有其他的位点。
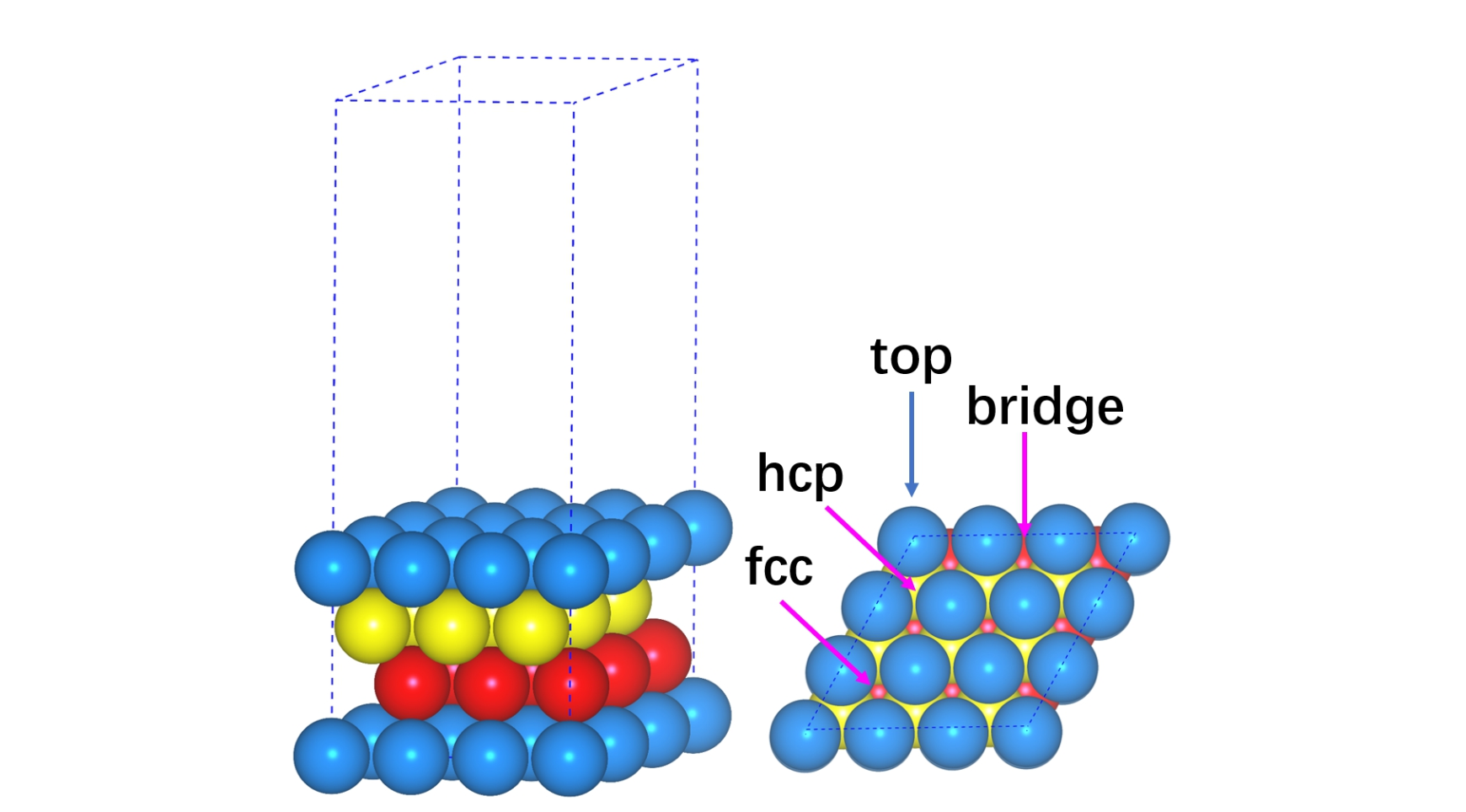
观察不同位点的结构特征:
在面心立方金属的(111)表面上有4种不同的位点: 1)Top位,前面已经花了很多时间介绍了,本节就不再啰嗦 2)Bridge位:从名字大家就可以推断这个位点的吸附为两个top原子的中间。 3)Fcc位和hcp位:这两个吸附位点都是在三个原子的中心,为hollow位。
那么这两个位点怎么区分呢? 如果表面的Hollow位正下方(即第二层,图中黄色的)有原子,为hcp位,在第三层的话(图中红色的),则这个hollow为fcc位。 由于fcc位点的原子在第三层,离表面最远(far),大家把英文单词的far 和 fcc位的第一个字母关联起来即可。远的那个就是fcc,近的是hcp。
怎么搭建bridge和fcc,以及hcp位的吸附模型? 1)Bridge位: 这个位点在两个原子的中心。设两个原子的坐标分别为(x1,y1, z1)和(x2, y2, z2),它们中心得坐标为: (x1+x2)/ 2, (y1+y2)/ 2, (z1+z2)
在这里,z1和z2 是相等的。所以我们可以通过两个原子的坐标,计算一下中心的坐标,然后按照之前O原子搭建的步骤,添加O原子的Bridge位的坐标即可。
2)Fcc位和hcp位的话,就更容易了,前面我们知道它们正下方分别为第三层和第二层的原子。那么我们在这两层随便取个原子,x和y 方向的坐标就确定了。
3)关于z方向的坐标,大家可以想象一下,如果O原子从top位移到Bridge,fcc和hcp,由于和O成键的原子多了,也就是O和表面结合地更强了,那么它在z方向的坐标肯定比在top位上要小。所以在这三个构型的z坐标,我们可以在top位O的基础上稍微调小一点。那么小多少呢? 一般来说0.1-0.2Å就可以了。
(后面Chapter 9快速解决)
在进行下面的快速编辑模型的内容,先给大家介绍一个linux下面常用的比较不同的命令:diff
diff[参数][文件1或目录1][文件2或目录2]
diff log2014.log log2013.log
如:“3c3”和“8c8”表示log2014.log和log20143log文件在3行和第8行内容有所不同;"11,12d10"表示第一个文件比第二个文件多了第11和12行。
diff 的normal 显示格式有三种提示:
a - add
c - change
d - delete
对于复杂的模型初筛:为了获取最稳定的结构,我们需要将这些可能的结构都优化一遍,然后通过吸附能来判断。但是这12个结构,我们都要优化的话,需要很多的机时,尤其是对于那些不太富裕的课题组来说,计算量着实不小。那么我们应该怎么办呢?
这里我们可以选择的办法有N个:主要是这两个:1) 减小K点; 2)减小模型。
1) 减小K点的做法 i)固定住所有的表面原子(H O分子除外) ii)使用gamma点进行计算
2) 减小模型的做法 i)删去底部的2层原子 ii)固定表层的所有原子 iii)正常优化进行计算
批量查看能量信息:
for i in */*; do echo $i $(grep ' without' $i/OUTCAR | tail -n 1| awk '{print $7}') ; done加排序:
for i in */*; do echo $i $(grep ' without' $i/OUTCAR | tail -n 1| awk '{print $7}') ; done | sort -k 2进一步优化:
到现在位置,前面的12个初始结构,可以快速的被筛选成了3个。将这三个结构复制step2的文件夹中。在此基础上,开始正常计算:
1) 将CONTCAR批量复制成POSCAR:
for i in *; do mv $i/CONTCAR $i/POSCAR ; done2) KPOINTS变回原来的(4x4x1): sed 批量操作
3) 表面两层原子放开: sed 批量操作
4) 默背一遍提交任务的几个输入文件:INCAR,KPOINTS,POSCAR,POTCAR,脚本,集中注意力思考是否还有没有考虑到的参数或者细节,确保无误后
5) 批量提交任务: qsuball.sh
6)等待结束,查看结果
结构细节:化学上,我们对键长,键角这些信息一定不要放过。测量一下Cu—O 的距离
计算吸附能: E_ads = E(slab+H2O) – E(slab) – E(H2O_gas)
表面吸附物种的频率计算 :
IBRION = 5 POTIM = 0.015 或者0.01-0.02范围的值都可,闲的没事也可以测试下。 NFREE = 2 NSW = 1 1-10000的值均可,没事也可以测试下。
POSCAR: 将表面的原子固定住
KPOINTS,一般来说,很多人都使用和优化过程一样的KPOINTS,但从我个人的经验来看,使用更小的K点来计算则是一个更加有效,节约机时的做法。 (111所花的时间也仅仅是441的一个零头。如果你是个糙哥,111的结果完全够用。如果你是个细心的软男,不敢用111,但又害怕浪费机时,221则是个很好地选择 )
只提取虚频,使用 grep f/i OUTCAR
EDIFFG 从1E-5到1E-7,振动频率变化很小。这个给我们的启示是,1E-5够用了
表面化学中,熵的贡献主要在吸附/脱附的阶段,因为分子损失/获得了大部分的平动和转动,对于表面吸附物种的熵: i)3N-6的振动部分贡献很小,可以忽略不计; ii)Frustrated rotation 大约在200-400cm 左右,也可以和振动部分一起忽略; iii)Frustrated translation部分,可以近似为二维平动来处理。也可以忽略,一般来说:忽略 是大家普遍的做法。如果理论基础不扎实,只想简单算算,那么你的腰椎间盘还是不要突出为妙。 iv)表面吸附位点的数目对熵的贡献,基本也可以忽略。
而在表面反应的阶段,我们计算反应热和活化能的时候通过下面的公式: ΔE = E(FS) – E(IS); Ea = E(TS) – E(IS) 这两个量的计算都是通过相减来得到的,而这个相减的过程中: i)大部分熵的贡献可以被抵消掉了; ii)小波数范围内零点能的矫正部分也会被抵消掉,所以零点能校正的时候,大胆地直接地把非虚频的部分直接加起来就可以了。 iii)如果你不信,那么可以通过频率计算一下IS,FS,或者TS的熵,然后计算矫正过的ΔE, Ea,会发现熵的贡献很小很小,所以一般大家都直接考虑零点能,而不考虑熵的贡献,如果有审稿人问你这个问题,用脚本算一下,回答审稿人说影响不大就是了。