Lammps Tutorial
Chapter 1 Lammps cookbook
此部分主要参考了Michael博客所记述的命令(renaissance😀),包含本人的大量补充
Installation:
最简单的安装方法(for ubuntu system)
x#pre-build fftw3 and OpenMPI#use apt-getsudo apt-get install fftw3-dev#add source-repostorysudo add-apt-repository ppa:gladky-anton/lammpssudo apt-get update#auto installationsudo apt-get install lammps-daily#testlmp_daily -in in.lj#auto updatesudo apt-get update#get a copy of the current documentation and examplessudo apt-get install lammps-daily-doc#uninstallsudo apt-get remove lammps-daily自己编译(有可能遇到各种各样的问题):
- 编译最简单单核单机版:
- 编译串行版(或开放其他自带选项的版本):
- 编译含有外界工具接口的版本:
Input and output file:
Input:在大部分模拟中的输入文件均为.in文件
in文件的写法遵循以下两条原则:#注释 回车换行 &连接两行,关键字+数值 定义命令
例如:
# LJ 12-6 system with NVE ensembleunits lj#units 关键字用来说明整个模拟体系所涉及到的物理量的单位制,后面的参数 lj 是在告诉 LAMMPS 所有物理量都采用无量纲的约化单位。LAMMPS 还有很多种单位制可以选择,这需要根据实际体系来具体考虑,例如“units si”、“units real”等等。atom_style atomic#atom_style 关键字用来说明模拟里面的基本物质单元所具备的属性,参数 atomic 表示所有的单元都是粗粒化的小球,如果是参数 angle 则表明每一个单元里面还包括了键长和键角这两个属性,参数 charge 表示每一个单元是粗粒化的小球另外还附有电荷。#这 4 条指令的组合用来构造模拟所需要的初始构型,在这里是按照晶格的方式创建原子。lattice fcc 0.8442# 第一条指令是说所有的原子的初始位置按照面心立方(FCC)的形式排列,晶格常数为 0.8842;region box block 0 5 0 5 0 5# 第二条指令是说模拟的空间区域在 X, Y 和 Z 三个方向上都是有0到5这个范围,注意这里的长度都是无量纲的create_box 1 box# 第三条指令是说按照第二条指令指定的空间区域创造一个模拟盒子,所有的原子都只能在这个模拟盒子里create_atoms 1 box# 第四条指令是说按照第一条和第二条指令指定的参数创造一种布满整个模拟盒子的原子,同时所创造的原子的种类也定义为数字“1”mass 1 1.0#mass 关键字用来给所有属于同一种类的原子设定质量的数值,后面跟着的第一个参数“1”是原子的种类,既要为所有种类为“1”的原子设定质量,第二个参数“1.0”才是质量的数值,这个指令的意思就是所有种类为“1”的原子的质量都是 1.0。velocity all create 3.0 825577 dist gaussian#velocity 关键字用来给原子设定初始速度,后面的参数 all 表示模拟盒子里所有的原子,不分种类都按照同一种方式设定初始速度。后面的一串参数是说所有的速度都是从满足高斯分布的随机数中抽取获得的。#这两条指令组合起来规定了原子之间的相互作用的形式pair_style lj/cut 2.5#第一条指令是说相互作用为LJ 12-6 势,截断半径为 2.5pair_coeff * * 1.0 1.0 2.5#第二条是说 LJ 12-6 中的 ε 和 σ 分别为 1.0 和 1.0,截断半径为 2.5。其实截断半径可以只声明一次,不需要两条语句都重复声明。neighbor 0.3 binneigh_modify every 20 delay 0 check no#这两条指令组合起来规定了 LAMMPS 如何构建 neighbor listfix 1 all nve#这条指令的意思是模拟在微正则系宗 NVE 下运行。timestep 0.005#这条指令的意思是模拟的步长 δt = 0.005。thermo 1000#每隔 1000 步模拟步长输出一次系统的热力学参量。run 10000#模拟运行 10000 步。clear输入文件结构:四大部分——Initialization、atom definition、settings i、run a simulation
Initialization:
xxxxxxxxxxunits lj/real/metal 三种单位系统dimensions 2d/3d(default)boundary 周期性/自由边界atom_style 原子属性(与力场参数中的atom_type不同)pair_style, bond_style, angle_style,dihedral_style, improper_style告诉使用何种力场Atom definition:
xxxxxxxxxxread_data 从data文件读入data文件可从CHARMM、AMBER、MSI转化(在tools文件夹)read_restart 从restart文件读入lattice region create_box create_atoms 创建原子replicate 已经设置好的原子可通过此命令生成一个更大规模的Settings:前面完成拓扑信息设置后,可在此部分设置一系列的:力场系数/模拟参数/输出选项
力场系数:pair_coeff, bond_coeff, angle_coeff, dihedral_coeff, improper_coeff, kspace_style, dielectric, special_bonds (此部分系数也可在data文件中指定)pair_styles在doc/pair.html查阅模拟参数:neighbor, neigh_modify, group, timestep, reset_timestep,run_style, min_style, min_modify,fix (最为重要,包含众多子命令,可施加一系列的边界条件、时间积分、诊断选项)规定lammps进行何种计算:compute、compute_modify、variable输出选项:thermo、dump、restart (dump, dump_modify, restart, thermo, thermo_modify, thermo_style, undump, write_restart)Run:
x
run 进行分子动力学模拟minimize 实施能量最小化temper 实施parallel tempering(replica-exchange) simulationdelete_atoms, delete_bonds, displace_atoms, displace_boxnewton arg
in文件运行遵循:从上到下依次执行的顺序,后面的所有操作均会依照前面的定义来(可当作一门程序语言理解)
第三方写in文件的程序:
- VMD TopoTools (Axel Kohlmeyer)
- Avogadro
- Packmol (见Chapter 2)
output:
肯定有的文件:
log.lammps——整个计算过程所有文件
与dump命令有关的输出——输出应力、能量、原子位置、速度
restart文件——write_restart命令
输入文件处理思路:
- 模型 → cif 格式 → vasp 格式 → lammps 格式
- 当然最简单的是用
open babeloravogadro直接转化 - 按传统的思路需要经过Materials Studio, VESTA, poscar2lammps 脚本
输出文件处理思路:
lammps 输出 cfg 格式 →pdb 格式 → 导入 ms 建模
dump 1 all cfg 100 dump.snap.*.cfg id type xs ys zs有时需要辅助
dump_modify命令,用来设置其中的元素类型cfg 格式是李巨提出来的,他也提供了 cfg 转换成 pdb 的脚本
cfg2pdb input.cfg output.pdb
Common Initialization Command Mannul :
units command:
units lj/real/metal/si/cgs/electron用于设置出现的所有的变量单位(除了lj为无量纲量单位外,其余的单位均遵循www.physics.nist.gov 上的规定)
real metal si cgs electron mass g/mol g/mol kg g atomic mass unit distance Å Å m cm Bohr time fs ps s s fs energy kcal/mol eV J ergs Ha velocity Å/fs Å/ps m/s cm/s 1.03275e-15 s force kcal/(mol Å) eV/Å N Dy Ha/Bohr torque kcal/mol eV N*m Dy*cm temperature K K K K K pressure atm bars Pa Dy/cm2 Pa dynamic viscosity Po Po Pa*s Po charge e e C esu e dipole e*Å e*Å C*m statcoul-cm Debye electric field V/Å V/Å V/m Dy/esu V/cm density g/cm3 g/cm3 kg/m3 g/cm3 不同的单位类型,也设置了默认的时间步和领域表层距离 :
lj real metal si cgs electron dt 0.005 tau 1 fs 0.001 ps 1e-8 s 1e-8 s 0.001 fs skin 0.3 sigma 2 Å 2 Å 0.001 m 0.1 cm 2 Bohr 该命令必须在模拟盒子被定义(使用命令 read_data 或 create_box)前使用(in文件之首)
dimensions command:
该命令用来定义模拟的维度。默认情况下, LAMMPS 运行三维模拟。如果要运行二维的模拟,需要在建立模拟盒子(使用命令 create_box 或 read_data)之前,先使用该命令进行设置。重启动文件也会保存该设置。
详细说明
dimension NN=2/3注意事项
注意: LAMMPS 中的有些模型将粒子作为有限尺寸的球体或椭球体对待,而不是点粒子。在二维模拟时,这些粒子仍然是球体或椭球体,而不是圆或椭圆,这也就意味着它们的惯性矩与三维情形下是相同的
boundary command:
设置模拟盒子沿着各个方向的边界条件。
单独的一个字母会将模拟盒子沿着某个方向的两个面设置为一样的边界条件。两个字母会将这两个面分别设置为不同的边界条件。模拟盒子的初始尺寸是由命令 read_data, read_restart 或 create_box 命令 设置的 。
详细说明
boundary x y zx,y,z 可取 p/s/f/m 中的一个字母或两个字母的组合。 其中:
p:周期性边界条件 periodic
p 代表周期性边界条件,就是说原子在跨越模拟盒子的边界时,会从盒子的另外一边再进入盒子里。设置为周期性的方向的盒子尺寸是可以通过常压边界条件或盒子变形(参考命令 fix npt 和 fix deform)而发生改变的。 p 必须同时用在某个方向的两个面上 。
f:非周期性固定边界条件 fixed
f 是将所对应的面设置为固定的。如果原子从这个面移动出去了,那么这个原子就丢失了。
s:非周期性包覆边界条件 shrink-wrapped
s 是将所对应的面设置为浮动的,不论原子在那个方向上移动到哪里,都会通过调整所对应的面的位置而将原子包围在盒子里。
m:非周期性包覆最小值边界条件 minimum value
m 是在 s 的基础上定义的,虽然包覆原子的行为会发生,但却被限制在一定的范围内。这个限制值是通过数据文件或重启动文件或命令 create_box 设置的。举例说明:如果在数据文件中设置了 z 的正方向为 50.0,那么即便模拟盒子在 z 的正方向上的最大值变的比 50.0 小, z 正方向上的这个面也只会在 50.0 或者其上的位置上。
注意事项:
对于非正交模拟盒子,如果倾斜因子的第二个维度(比如 xy 的 y 方向)是周期性的,那么在执行周期性的时候会强制倾斜因子带来的偏移。如果第一个维度是包覆型的(即 s 或 m),那么包覆是针对倾斜面而言的。举例来说,对于一个正的 xy倾斜因子,模拟盒子的 xlo 面和 xhi 面就是沿着+y 方向进行倾斜的面。这些倾斜面决定了模拟盒子在 x 方向的范围,原子也是被包覆在这些倾斜面内。
对于二维模拟来说, z 必须设置为周期性
atom_style command:
定义模拟过程中原子的类型,它会决定原子包括哪些属性。该命令必须在建立模拟盒子(使用命令
read_data或read_restart或create_box)之前使用。一定原子的类型被设置了,在模拟过程中就不能在改变了,因此尽量更加通用的类型,以免某些属性没有被包括却被用到了。举例来说,对于 bond 类型,原子没有角度项,也不能在模拟的过程中为模型添加角度属性。尽量使用更加通用的类型,虽然这可能会因为某些属性没有用到,同时会稍微降低效率,但总比出错好。 类型的选择会影响每个原子所能够存储的性质、在计算力的时候处理器间能够交换的性质以及在数据文件中所能够显示并被命令
read_data读入的性质。详细说明
atom_style style paras其中 :
style=angle/atomic/body/bond/charge/dipole/electron/ellipsoid/full/line/meso/molecular/peri/sphere/tri/hybrid只有
body和hybrid需要设置paras(具体参考原文档)各参数详解:
style properties system anglebonds and angles 有刚度的高分子(bead-spring) atomiconly the default values 粗粒化液体/固体/金属 bodymass, inertia moments, quaternion, angular momentum 任意物体(由后续参数指定) bondbonds 高分子(bead-spring) chargecharge 带电荷的原子系统 dipolecharge and dipole moment 含有偶极粒子的系统 electroncharge and spin and eradius 电子力场(3d) ellipsoidshape, quaternion, angular momentum 非球形粒子(粒子是椭球体,每个粒子存有一个标记,用来区分该粒子是有限尺寸的椭球还是点粒子。如果是椭球,那么每个粒子就存有形状矢量,这个矢量包括椭球体的三个直径和一个代表其方位的四维矢量) fullmolecular + charge 生物分子 lineend points, angular velocity 刚体(颗粒是理想化的线段) mesorho, e, cv SPH颗粒(光滑粒子流体动力学计算) moleculebonds, angles, dihedrals, impropers 不带电荷的分子 perimass, volume 介观动力学模型(粒子是球形的,每个粒子存有其质量和体积) spherediameter, mass, angular velocity 颗粒化模型 (粒子是球形的,每个粒子存有其直径和质量。如果其直径大于 0,粒子是有限尺寸的球;如果直径等于 0,它是一个点粒子(point particle)) tricorner points, angular momentum 刚体(颗粒是理想化的平面三角形) wavepacketcharge, spin, eradius, etag, cs_re, cs_im AWPMD (Antisymmetrized Wave Packet Molecular Dynamics model)(与类型 electron 类似,但是电子由几个按coefficients cs= (cs_re,cs_im)参数加在一起的Gaussian波包组成。 每个波包在lammps里面作为分开的颗粒处理, 属于相同电子的波包必须有相同的etag值) 应用例
xxxxxxxxxxatom_style atomicatom_style bondatom_style fullatom_style body nparticle 2 10atom_style hybrid charge bondatom_style hybrid charge body nparticle 2 5注意项
注意 1:
通过命令
fix property/atom对原子添加它原先没有的属性是可以的,比如分子 ID。该命令也允许为原子添加新的由整数或浮点数组成的自定义属性。参考命令fix property/atom,了解更多细节和例子。上面的类型中,除了sphere/ellipsoid/electron/peri/wavepacket/line/tri/body定义的是有限尺寸粒子,其他的类型定义的都是点粒子。注意 2:
一般来说,模拟时需要单独一种的原子类型(non-hybrid)。但如果模拟中的有些原子不需要某些性质,但其他的原子需要,那么就要使用包含所有这些性质的类型。比如,如果有些原子需要电荷,而另外一些不需要,要使用类型
charge;如果有些原子有键,而另外一些没有,要使用类型 bond。 唯一可能用到类型hybrid的情况是没有任何一种类型包括了所有需要的性质。比如,如果你要使用极性粒子,并且要这些粒子在扭矩下旋转,那么你就需要用到atom_style hybrid sphere dipole。如果使用类型hybrid,那么原子就会储存由这两种类型定义的所有性质。该命令不能在模拟盒子被定义(使用命令
read_data或create_box)之后使用。注意 3:
类型
angle/bond/full/molecular是 MOLECULAR 包的一部分;类型line/tri是ASPHERE 包的一部分;类型body是 BODY 包的一部分;类型dipole是 DIPOLE包的一部分;类型peri是 PERI 包的一部分;类型electron是 USER-EFF 包的一部分;类型 meso 是 USER-SPH 包的一部分; wavepacket 是 USER-AWPMD 包的一部分。只有在 LAMMPS 编译的时候把相应的包编译进去了,这些类型才可以使用。pair_style command:
bond_style command:
angle_style command:
Common Atom definition Command Mannul :
read_data command:
read_restart command:
该命令用来读入之前的模拟过程保存下的重启动文件。该命令可以帮助你实现接着之前的模拟过程继续进行。下面会告诉你哪些信息被存储在了重启动文件中。
重启动文件是二进制格式的,它是用来精确重启动之前模拟过程,就是说重启动之后的模拟会精确的接着之前已经进行的模拟继续进行。但也有一些因素会因为舍入误差而影响到这种重启动的精确性,这种情况下这两个模拟间就会有一些偏差。
这些影响因素包括使用数量不同的处理器进行计算,或者改变了某些设置,比如那些与命令
newton或processors相关的设置。 LAMMPS会在这种情况下给出警告。有些
fix命令不能被重启动,尽管它们会提供相近的统计结果。这种命令包括fix shake和fix langevin。有些势类型不能被重启动,尽管它们会提供相近的统计结果。这是因为力的计算是依赖于原子速度的,而在每个时间步计算力的时候,使用的是半个时间步的速度值。当重启动一个计算时,力的计算却被初始化为全步的速度值,这就与写入重启动文件时的计算直接进行下去的结果不同。这些势类型有
granular pair styles,pair dpd和pair lubricate。如果你在重启动一个计算时,从一开始就与产生重启动文件的计算有了不一样,这应该是 LAMMPS 的 bug,你可以报告给 LAMMPS 软件。
因为重启动文件是二进制的,所以这些文件一般来说是不能拷贝到其他的机器使用的,但你可以使用工具 restart2data 将其转换成文本文件。
与写重启动文件(参考命令 write_restart 和 restart)类似,读入的重启动文件的文件名也可以包括两种通配符。
如果是符号“”,那么会在当前目录下匹配所有文件名中将“*”替换成时间步的文件,但只有最大时间步的那个文件会被读入。也就是说,最后写入的那个重启动文件会被读入用于重启动模拟。如果你想让你的计算从之前停下来的地方开始,那么这种方式非常实用。参考命令
run和它的upto选项,了解怎样设置run命令可以不需要在新的计算中更改命令 run 的设置。如果是符号“%”, LAMMPS 会要求有一系列的重启动文件存在。在命令
restart和write_restart命令中,已经介绍了这一些列重启动文件是如何创建的。read_restart命令首先会读入将“%”替换为 base 的那个文件,该文件告诉 LAMMPS 之前的模拟设置了多少个处理器。然后read_restart命令会读入剩下的那些重启动文件。举例来说,如果在写入的时候,设置重启动文件的文件名为 save.%,那么在使用read_restart读入的时候,就会读入这些文件 save.base, save.0, save.1, ……, save.P-1(这里面 P 是创建重启动文件时处理器的数量)。在当前的 LAMMPS 模拟中,所有的处理器同时读入这些文件,每个处理器会读入文件数量大致相等。创建重启动文件的处理器数量可以跟读入重启动文件的处理器数量不一样。对于支持并行 I/O 的并行机来说,这是一种快速的输入方式。对一个模拟而言,下列信息会被存储在了重启动文件中:单位样式 units、原子样式 atom style、模拟盒子的尺寸和形状、边界条件、定义的组、与原子类型相关的设置(如质量)、与每个原子相关性质(包括它们被分配到的组和分子拓扑性质)、势函数类型及参数、特殊键 specail_bonds 的设置。这就意味着与这些量相关的命令不需要在读入重启动文件中的输入脚本中再重新进行指定。当然,你也可以重新指定这些设置。
有一个例外是有些势类型的信息没有存储在重启动文件中。具体哪种类型,会在介绍那种势类型的时候专门指出来。命令 bond_style hybrid (and angle_style,dihedral_style, improper_style hybrid) 也是这样的。
所有由命令
pair_modify所指定的设置,比如 shift 和 tail,会与势类型一起存储在重启动文件中。但命令pair_modify compute是唯一的例外。命令
kspace_style的相关信息没有存储在重启动文件中,所以如果你希望使用Ewald or PPPM 求解器,那么这些命令还需要在重启动文件被读入后重新指定。 所有与fix相关的命令都没有存储在重启动文件中。这就意味着在新的输入脚本中需要重新指定所有用到的 fix 命令。需要注意的是,有一些 fix 命令会将它们内在的状态写入到重启动文件中,这就使得在开始重启动模拟之后,这些 fix 命令可以继续起作用,但这些却需要被激活。你只需要在新的输入脚本中使用与写入重启动文件的那个脚本相同的 fix-ID 和 group-ID 来定义这些 fix 命令,就可以将它们激活。如果 fix 命令能够匹配上,那么 LAMMPS 会输出一个信息,指示这个 fix 命令被重新激活了。如果在执行新输入脚本中计算或能量最小化之前没有发现可以匹配的 fix 命令,那么从重启动文件保存的 fix 命令的内在状态信息就会被丢弃掉。参考fix命令,了解具体是哪个具有这样的重启动方式。使用命令
fix_shake或delete_bonds关掉的键作用,在被写入到重启动文件时,会被打开。所以,在读入重启动文件后,需要重新将这些键作用关掉。断掉的键也被写入到了重启动文件中(断键用类型 0 表示),因此当读入重启动文件的时候,这些键仍然是断掉的。
详细说明:
read_restart filefile:要读入的二进制重启动文件的文件名应用例:
xxxxxxxxxxread_restart save.10000read_restart restart.*read_restart poly.*.%注意项:
除了上面列举的信息之外,其他任何信息都没有被存储在重启动文件中。这就是说,对于读入重启动文件的输入脚本来说,需要指定的量有:时间步长
timestep、热力学信息thermo_style、领域列表neighbor、领域列表设置neigh_modify、dump文件输出、几何区域region等lattice command:
region command:
该命令用于定义一个空间几何区域。很多其他命令都会使用该命令定义的区域。举例来说,
create_atoms可以在定义区域中创建原子,create_box命令可以根据定义区域定义模拟盒子,group命令可以将定义区域中包括的原子定义为一个组,delete_atoms命令可以将定义区域中的原子删掉,fix wall/region可以将定义区域的表面定义为壁面(boundary wall)。详细说明:
region regionID style args keyword arg ...regionID-待定义区域的 IDstyle=delete / block / cone / cylinder / plane / prism / sphere / union / intersectdelete: no args 删除指定的区域。因为定义冗余的区域并不会占用太大的开销,所以一般不需要这样做,除非在你的输入脚本中定义了大量了区域。
block
args = xlo xhi ylo yhi zlo zhixlo,xhi,ylo,yhi,zlo,zhi = 各个维度上的范围值cone
args = dim c1 c2 radlo radhi lo hidim = x or y or z圆锥台轴线方向;c1,c2圆锥台轴线在另外两个维度上的坐标值 ;radlo,radhi圆锥台上下底面的半径;lo,hi圆锥台轴线方向的范围值;底面半径必须定义得不同,两个底面半径都可以设置为0,但不能同时设为 0.cylinder
args = dim c1 c2 radius lo hiradius圆柱体半径,可以使用变量 余同coneplane
args = px py pz nx ny nzpx,py,pz平面上一个点的坐标;nx,ny,nz平面法矢坐标prism
args = xlo xhi ylo yhi zlo zhi xy xz yzxlo,xhi,ylo,yhi,zlo,zhi平行六面体在各个维度上的范围值ijj 方向在 i 方向的倾斜值 (i,j = x,y,z; i ≠ j)如果要使用
create_box命令,并以已经定义为 prism 类型的区域为范围定义三斜式的模拟盒子,那么倾斜因子(xy,xz,yz)不能超过各个维度上对应盒子长度的一半。举个例子,如果 xlo=2, xhi=12,那么模拟盒子的长度就是 10,这也就要求 xy倾斜因子必须在-5 到 5 之间。类似地,倾斜因子 xz 和 yz 必须在-(xhi-xlo)/2 和+(yhi-ylo)/2 之间。需要注意的是,这并不是一个限制,因为如果最大的倾斜因子应该是 5,那么将其设置为-15, -5, 5, 15, 25……在几何上都是等效的。sphere
args = x y z radiusx,y,z球心坐标;radius球体半径,可以使用变量block / cone / cylinder / prism 类型中参数 lo/hi 的值可以指定为 EDGE 或INF。
- EDGE 是指所定义方向一直延伸到所在方向模拟盒子的边界。需要注意的是,这里所说模拟盒子的边界是指当前盒子的边界。换句话说,如果模拟盒子在模拟的过程中发生了改变,按着这种方式定义的区域并不会随着改变。
- INF 是指一个非常大的负数或正数(1.0e20),所以即便模拟盒子在模拟过程中发生改变了,使用该定义仍然能将其包括到区域中。
- 如果模拟盒子还没有创建,那么在使用 region 定义区域的时候就不能使用 EDGE或 INF(译注:这是显然的,因为 EDGE 和 INF 都是相对于模拟盒子来说的)
- 对于 prism 类型的区域,如果某个倾斜因子不为 0,那么它所对应的两个维度上lo/hi 都不能设为 INF。举例来说,如果 xy 倾斜因子设为非零,那么 xlo/xhi 和ylo/yhi 都不能设为INF。
union
args = N regionID1 regionID2 ...N要进行并集操作的区域的数目,必须大于等于 2regionID1,regionID2, ...要进行并集操作的区域的 IDintersect
args = N reg-ID1 reg-ID2 ...N要进行交集操作的区域的数目,必须大于等于 2
keyword=side/units/move/rotatesidevalue = in/outin指定几何体内侧作为区域out指定几何体外侧作为区域通过将该关键字与
union和intersect类型配合使用,可以构建复杂的几何区域。举个例子,如 果将两个球体的内部分别定义成了区域,并对这两个区域使用类型为union side=out的操作,那么所定义的区域就是整个模拟盒子之内、除了两个球体之外的所有空间unitsvalue = lattice/box以晶格距离/模拟盒子作为几何距离单位若取值 lattice,坐标是以晶格距离为单位的,但前提是必须已经使用 lattice 命令定义了晶格距离。具体到不同类型,在不同维度上是不同的,如下:
block类型: xlo/xhi 是以 x 方向的晶格距离为单位。类似地, ylo/yhi 和 zlo/zhi 分别是以 y 和 z 方向的晶格距离为单位。cone类型: lo/hi 是以 dim 所在方向的晶格距离为单位。 c1/c2 则分别以另外两个径向维度方向上的晶格距离为单位。圆锥台上下底面的半径是以与 c1 对应的那个方向上的晶格距离为单位。cylinder类型: lo/hi 是以 dim 所在方向的晶格距离为单位。 c1/c2 则分别以另外两个维度方向上的晶格距离为单位。圆柱体半径是以与 c1 对应的那个方向上的晶格距离为单位。plane类型: px 和 nx 是以 x 方向的晶格距离为单位。类似地, py/ny 是以 y 方向的晶格距离为单位, pz/nz 是以 z 方向的晶格距离为单位。prism类型: xlo/xhi 是以 x 方向的晶格距离为单位。类似地, ylo/yhi 和 zlo/zhi 分别是以 y 和 z 方向的晶格距离为单位。 xy 和 xz 是以 x 方向的晶格距离为单位, yz 是以 y 方向的晶格距离为单位sphere类型:球形坐标 x,y,z 分别是以 x,y,z 方向的晶格距离为单位,球半径是以 x方向的晶格距离为单位
moveargs = v_x v_y v_zv_x,v_y,v_z = x, y, z 方向的位移量,equal 类型的变量rotateargs = v_theta Px Py Pz Rx Ry Rzv_theta旋转角, equal 类型的变量,弧度单位Px,Py,Pz旋转操作的中心点Rx,Ry,Rz旋转轴矢量如果使用
move或rotate关键字,那么所定义的区域就变成了动态的,也就是说它的位置和朝向是会随着时间发生变化。这两个关键字不能用于 union 或 intersect类型的区域。可用于定义一块区域位置随时间的变化:
xxxxxxxxxxvariable dx equal ramp(0,10)region 2 sphere 10.0 10.0 0.0 5 move v_dx NULL NULL定义一个摇晃的区域:
xxxxxxxxxxvariable dy equal swiggle(0,5,100)variable dysame equal 5*sin(2*PI*elaplong*dt/100)region 2 sphere 10.0 10.0 0.0 5 move NULL v_dy NULL
应用例:
xxxxxxxxxxregion 1 block -3.0 5.0 INF 10.0 INF INFregion 2 sphere 0.0 0.0 0.0 5 side outregion void cylinder y 2 3 5 -5.0 EDGE units boxregion 1 prism 0 10 0 10 0 10 2 0 0region outside union 4 side1 side2 side3 side4region 2 sphere 0.0 0.0 0.0 5 side out move v_left v_up NULL注意项:
注意 1: Regions in LAMMPS do not get wrapped across periodic boundaries, as specified by the
boundarycommand. For example, a spherical region that is defined so that it overlaps a periodic boundary is not treated as 2 half-spheres, one on either side of the simulation box.注意 2:不论是否用
dimension命令将模拟设为 2d 或 3d, LAMMPS 中所定义的 区域总是三维的。所以在进行 2d 模拟时,你需要仔细定义区域,以使其与 2d 的 x-y 平面相交,并产生正确的交集区域。注意 3:
union和intersect类型的区域依赖于产生它们的每一个子区域,因此在定义 union 或 intersect 类型的区域后,不能删除这些子区域create_box command:
create_atoms command:
使用介绍:
该命令用来在晶格阵点上创建原子,或创建一个单独的原子,或创建一些列随机原子。也可以用命令
read_data或read_restart通过直接给出原子坐标的方式创建原子。在使用该命令之前,模拟盒子必须是存在的(使用create_box命令创建),同时晶格也必须已经被定义(使用lattice命令)。但对于创建 single 类型且以 box 为单位的原子,或创建 random 类型的原子时,不需要先定义晶格。
该命令是向已经存在的体系中继续添加原子。换句话说,该命令可以多次使用,从而可以在模拟盒子中创建多组原子。举例来说,通过交错地使用
create_atoms命令和lattice命令(配置为不同晶向orientations),就可以创建晶界; 联合使用create_atoms命令和delete_atoms命令,可以创建非常复杂的体系。(?)create_atoms命令也可以在已经读入的体系中继续创建原子。在所有列举的这些情况中,都需要注意不要让新创建的原子与已经存在的原子重叠。可以使用delete_atoms命令来处理重叠原子的问题。详细说明:
create_atoms type style args keyword values ...type-要创建的原子类型(用1到N的数字代替)
style-
box/region/single/randombox:arg=none该命令在整个模拟盒子中所有的晶格阵点上创建原子。如果你的模拟盒子是周期性的,你应该确保其尺寸是晶格距离的整数倍,从而避免在盒子边界处可能存在的原子重叠。如果你的盒子是周期性的,并且在某个方向上盒子的尺寸是晶格距离的整数倍,那么 LAMMPS 会在边界上只放置一个原子(译注:边界上的原子不会丢失或在两个面同时出现)。
region:arg=regionID仅在region内的原子才会被创建该命令会在 region-ID 所指定的区域与模拟盒子相交的公共区域内创建原子。参考命令
region,可以了解更多细节。需要注意的是,这里定义的区域可以在模拟盒子内,也可以在模拟盒子外。还需要注意,即便你在这里定义的区域与周期性模拟盒子的尺寸是一样的, LAMMPS 所执行的逻辑跟 box 类型也不一样,就是说并不能像 box 类型那样确保在边界上只有一个原子。所以如果你希望达成像 box 类型那样的效果,你最好使用 box 类型,或者就是非常精确地调整region 的尺寸来获得你想要的原子。single:args=x y zx,y,z为创建原子的坐标将指定坐标的原子添加到系统中。这对于调试或者创建一些列手动添加的原子会比较有用
random:args=N seed regionIDN-要创建的原子数seed-随机数,用作种子regionID-在region内创建原子,如果设置为NULL,则在整个模拟盒子内创建在系统中按着随机坐标产生 N 各原子,这对于产生无定形系统会比较有用。根据指定的随机种子数 seed,程序会依次创建 N 个随机原子。这 N 个随机原子的坐标与所使用处理器的个数没有关系。另外,如果 region-ID 设置为NULL,那么创建的原子会随机填充模拟盒子的任何位置;如果指定了特定的region-ID,那么原子就只会随机填充在模拟盒子与 region-ID 所共同指定的区域。 注意:使用 random 类型产生的原子很有可能是相互重叠的(译注:或者是距离较近,不太符合实际物理),从而导致计算出较大的力或能量。因此,在你开始进行正常的动力学计算时,最好先使用
minimize对体系进行能量最小化,或先使用fix nve/limit进行动力学计算。keywords-
basis/remap/unitsbasis values=M itypeM哪个基本原子itype该基本原子的类型(1~N)用来为要创建的特定的基本原子指定原子类型。参考
lattice命令,了解如何为原胞定义基本原子。默认情况下,所有创建的原子都被指定为参数itype说指定的原子类型remap values=yes/no仅仅在
single类型下使用。如果将其设置为yes,那么当指定的位置在模拟盒子外面时,它会将其挪到盒子里面,就好像在相应的方向上设置了周期性边界条件一样。如果将其设置为 no,就不会进行类似上面的调整,就是说如果创建原子的位置在模拟盒子外面,那么就不会创建原子。units values=lattice/box以晶格距离/模拟盒子作为单位用来设定要创建的 single 类型的那个原子的坐标的单位。
若取值
box,坐标的单位就与命令units说指定的距离单位相同,比如units设为real或metal时,其单位为 Angstroms。若取值
lattice,坐标是以晶格距离为单位的。应用例:
create_atoms 3 region regsphere basis 2 3create_atoms 3 single 0 0 5注意事件:
所创建的原子的 ID 是遵从如下规律:所有待创建原子的 ID 是连续的,其中第一个被创建原子的 ID 是紧接着体系中已经存在原子的最大的 ID。使用不同数目的处理器进行计算时,不能保证某个特定的原子会分配到同样的 ID。 除了原子的 ID 之外,原子类型、坐标以及其他性质都设为默认值。具体到有哪些性质,是由
atom style决定的。参考set和velocity命令,了解如何改变这些默认值。sphere类型会将默认的粒子直径和密度设置为 1.0,那么粒子对应的质量就不是1.0,而是 PI/6 * diameter^3 = 0.5236. ellipsoid类型会将默认的粒子形状设置为(0.0 0.0 0.0),密度设置为 1.0,也就是一个点粒子,而不是真正的 ellipsoid,并且质量为 1.0。 peri类型会将默认的体积和密度设置为 1.0,因此粒子的质量也就是 1.0。 可以使用set命令来重写这些默认设置。mass:
为某一种或几种类型的原子设置质量。每种类型原子的质量也可以在 data file 文件中使用
Masses关键字设置,并由read_data命令读入。参考units命令,了解不同单位制下质量的单位在 data file 文件中,使用
Masses关键字设置原子类型的质量与输入脚本中使用mass命令类似,区别在于不能使用星号通配符。举例来说,应用例的第一个例子在data file 文件中设置时,只需要在 Masses 关键字下面设置如下的文本:1 1.0范式:
mass I valueI原子类型索引
I可以使用下面两种方式中的任意一种进行设置:显式地设置为某个数值,为某种特定类型的原子设置质量,如下面的第一个例子。
使用星号通配符,同时为多种类型的原子设置质量。可以接受的通配符样式有:
“*” or “*n” or “n*” or “m*n”。如果用 N 表示原子类型的数量,那么:- 只使用星号,即
*,就表示从 1 到 N 的所有原子类型。 - 以星号开头的样式,如
*n,就表示从 1 到 n(包括在内)的所有原子类型。 - 以星号结尾的样式,如
n*,就表示从 n 到 N(包括在内)的所有原子类型。 - 星号在中间的样式,如
m*n,就表示从 m 到 n(包括在内)的所有原子类型。
- 只使用星号,即
value质量值
应用例:
xxxxxxxxxxmass 1 1.0mass * 62.5mass 2* 62.5注意事项:
需要注意的是,只有当
atom_style命令定义的原子类型有类原子质量(per-type atom mass)属性时,才需要使用mass命令来定义质量。目前来说,除了sphere,ellipsoid,peri之外,其他所有的类型都需要定义原子质量。对于这些特殊的类型,需要为每一个粒子设置质量。单原子质量可以通过read_data命令从 data file文件中读入,也可以使用create_atoms命令设置为默认值,还可以通过set mass或set density命令设置为新值。(译注:类原子质量指的是一类原子具有相同的质量,而单原子质量是指不同原子有不同的质量。)另外需要注意的是,
pair_style eam命令所使用的 EAM 势函数文件中有原子类型质量的设置,因此不需要再额外使用mass命令重复定义原子质量如果你使用包括一种或多种子类型的命令
hybrid atom style,那么你就需要同时定义每种类型的原子质量和子类型中每个原子的质量。程序在运行中,会忽略类原子质量,而使用单原子质量。只有在模拟盒子定义(
read_data, read_restart, create_box)好了之后,才能使用该命令在运行模拟之前,需要定义所有类型原子的质量。在使用命令
velocityorfix shake之前,也需要定义原子质量设置的质量值必须大于 0
replicate command:已经设置好的原子可通过此命令生成一个更大规模的
Common Setting Command Mannul :
力场系数部分:
- pair_coeff
- bond_coeff
- angle_coeff
- dihedral_coeff
- improper_coeff
- kspace_style
- dielectric
- special_bonds
模拟参数部分:
neighbor
neigh_modify
group
timestep
该命令为之后的分子动力学模拟设置时间步长。参考命令 units,了解时间步长的单位。默认的时间步长依赖于模拟中所采用的单位类型,参考下面的默认设置。 如果命令 run_style 设置为 respa,那么参数 dt 设置的是最外层循环的时间步长。
详细说明
timestep dt注意项
单位与
unit保持一致reset_timestep
run_style
min_style
该命令为
minimize命令选择一种能量最小化的算法。详细说明:
min_style stylestyle=cg/hftn/sd/quickmin/firecg类型是选择 Polak-Ribiere 版本的共轭梯度法 conjugate gradient (CG)。 At each iteration the force gradient is combined with the previous iteration information to compute a new search direction perpendicular (conjugate) to the previous search direction. The PR variant affects how the direction is chosen and how the CG method is restarted when it ceases to make progress. 对于绝大多数的问题来说, PR 版的共轭梯度算法都被认为是效果最好的。hftn类型是 Hessian-free truncated Newton 算法。 At each iteration a quadratic model of the energy potential is solved by a conjugate gradient inner iteration. The Hessian (second derivatives) of the energy is not formed directly, but approximated in each conjugate search direction by a finite difference directional derivative. When close to an energy minimum, the algorithm behaves like a Newton method and exhibits a quadratic convergence rate to high accuracy. 在大多数情况下, hftn 算法的跟 cg 算法效果相近,不过在 cg 算法出现问题时,可以考 虑更换选择 hftn 算法。 hftn 类型不受 min_modify 命令影响。sd类型是最速下降算法 steepest descent。在每个迭代步中,其搜索方向 is set to the downhill direction corresponding to the force vector (能量的负梯度方向)。一般来说, sd 算法都没有 cg 算法收敛快,但在某些特定的情形下可能会更加稳定。quickmin类型是 Sheppard 提出的一种阻尼动力学算法,其阻尼参数与速度矢量沿着每个原子当前力矢量上的投影相关。该类型在进行能量最小化之前把每个原子的速度被初始化为 0.0。fire类型是 Bitzek 提出的一种阻尼动力学算法,它与 quickmin 算法类似, bu adds a variable timestep and alters the projection operation to maintain components of the velocity non-parallel to the current force vector. 该类型在进行能量最小化之前把每个原子的速度被初始化为 0.0。注意项
quickmin算法和fire算法对于 NEB 计算(使用 neb 命令)会比较有用类型
quickmin和fire暂时还不支持fix box/relax命令,也不支持在 eFF 模型中包含电子半径[electron radius]的能量最小化。min_modify
fix为一组原子施加 fix 约束。在 LAMMPS 中, fix 是施加在分子动力学时间步或能量最小化过程中的某种操作。举例来说,它可能是在时间积分的过程中更新原子的位置和速度,或是控制温度,或是给原子施加约束力,或是强制某种边界条件,或计算过程诊断,等等。在 LAMMSP 中有一些可以使用的 fix 命令,具体参考后面的列表。
fix 命令会在时间步中的不同过程中执行相关的操作。如果有 2 个或多个 fix 命令作用于时间步中的同一过程,它们会按着在输入脚本中定义的顺序依次被激活。
fix 命令所施加的约束可以被
unfix命令删去。fix 命令会计算某种类型的量(全局量、单原子量或局部量),这些量可以被其他命令使用或输出 (一个单独的 fix 命令只可能会产生这三种类型量的某一种 )
- 全局量(global)是系统维度的量,比如体系的总能
- 单原子量(per-atom)是每个原子都具有的量,比如每个原子的位移矢量。不在 fix命令所约束的组内的原子的该单原子量被设为 0
- 局域量(local)是每个处理器基于它们所拥有的原子而计算出的量。原子可能有多个或没有局域量
- 全局量、单原子量和局域量都有三种存在形式:单独的标量、一维矢量和二维阵列。
如果要使用
fix命令计算出的量,比如使用输出命令将它们输出,可以使用下面的方括号记号来引用它们。其中的ID是 fix 命令的ID使用形式 含义 ID 整个标量、整个一维矢量、整个二维阵列 ID[i] 一维矢量中的一个元素、二维阵列中的一列 ID [i] [j] 二维阵列中的一个元素 在 LAMMPS 中, fix 命令产生的值有下列的使用方法 :
- 全局量可以使用命令
thermo_style customorfix ave/time输出,也可以以 equal 类型或 atom 类型的变量进行引用。 - 单原子量可以使用
dump customorfix ave/spatial命令进行输出,也可以使用fix ave/atom命令对时间进行平均,或使用compute reduce命令进行降维,或使用atom-style 类型的变量进行引用。 - 局域量可以使用
compute reduce命令进行降维,或者使用fix ave/histo命令进行直方图化。
fix 命令计算的全局量既可以是内部的,也可以是外部的。 “内部”是说其值独立于模拟中的原子数,比如温度。 “外部”是说其值的大小与模拟中的原子数有关系,比如总的转动动能。命令
Thermodynamic output会将外部量的值对体系中的原子数进行规范化,具体看“thermo_modify norm”的设置。但对于内部量,它不会进行规范化。如果使用其他方法对 fix 量进行引用,比如变量 variable,你需要了解它是内部量还是外部量详细说明
fix ID group-ID style argsxxxxxxxxxxID = fix 命令的 ID (ID 只能包含字母、数字和下划线)group-ID = 该 fix 命令所作用的原子组的 IDstyle = fix 类型名(最下有列表)args = 特定类型 fix 命令所需要的参数每一种类型的 fix 命令都有单独的文档页面来介绍其参数和它具体是做什么的。下面是 lammps 中可用的 fix 命令列表 (详细可见Chapter 2)
应用例
xxxxxxxxxxfix 1 all nvefix 3 all nvt temp 300.0 300.0 0.01fix mine top setforce 0.0 NULL 0.0注意项
注意 1:使用
unfix命令是唯一用来关闭 fix 命令所施加的约束的方法。如果只是指定一种新的类似的 fix 命令并不会关闭之前的 fix 命令。这对于进行积分的 fix 命令特别重要。举例来说,如果你先定义了fix nvt命令,然后又定义了fix nve命令,这样做并不会将先定义的 NVT 时间积分取消掉,而是这两个时间积分同时进行。 如果你在定义一个新的 fix 命令时,所使用的 ID 和类型名与某个已经定义的 fix 命令完全相同,那么先前定义的那个 fix 命令就会被删除掉,而新的 fix 命令会被创建,所使用的参数也是重新定义的。这样做就好像是在定义这个新的 fix 命令之前,先使用 unfix 命令取消了旧 fix 命令,除了说它们所作用的顺序会因着定义的位置不同而有所差异。另外需要注意的是,这种重新定义 fix 命令也会将先前使用
fix_modify命令定义的,与旧 fix 命令相关的其他任何改变都清除掉。 fix_modify 命令可以用来重置 fix 命令定义的某些设置。具体细节可以参考特定类型的 fix 命令。 有些 fix 命令会在写二进制重启动文件(restart 命令或 write_restart 命令)时保存其内部状态。这些 fix 命令会在重启动该模拟时继续对计算过程起作用。参考read_restart 命令,了解在读入重启动文件的输入脚本中,如何重新指定 fix 命令。参考具体类型的 fix 命令,了解哪些命令会被写入到重启动文件中。注意 2:可以使用 fix 量的命令和变量并不是可以使用任何一种存在形式的量,比如一个命令可能会需要矢量类型的量,而不是标量类型的。这也就意味着在引用 fix计算的量时不能含糊,而需要注意它的存在形式。这也会在具体介绍这些命令时详细的解释到。
fix_modify该命令用来修改过之前定义过的
fix命令的一个或多个参数。只有某些特定类型的fix命令支持修改参数。详细说明
fix_modify fix-ID keyword value ...x
fix-ID = 需要修改的 fix 命令的 ID可以添加 1 个或多个关键字keyword = temp/press/energytemp value = 计算温度的 compute 命令的 IDpress value = 计算压强的 compute 命令的 IDenergy value = yes or no- 关键字
temp决定 fix 命令如何计算温度。其所指定的 ID 是之前定义过的compute命令的 ID,而且必须是可以计算温度类型的compute命令。实际上,所有可以计算温度的 fix 命令里面已经默认定义了自己的compute方法。所以,如果你使用fix_modify命令时,它所定义的计算温度的方法就会覆盖 fix 命令里默认的计算方法。 - 关键字
press决定 fix 命令如何计算压强。其所指定的 ID 是之前定义过的compute命令的 ID,而且必须是可以计算压强类型的 compute 命令。实际上,所以可以计算压强的 fix 命令里面已经默认定义了自己的compute方法。所以,如果你使用fix_modify命令时,它所定义的的计算压强的方法就会覆盖 fix 命令里默认的计算方法。 - 有些对计算体系总势能有贡献的
fix命令,关键字energy会决定在进行热力学信息输出势能时,是否将该贡献值包括在其中。参考thermo_style命令,了解势能是如何输出的。另外,该贡献值本身可以通过在 thermo_style custom 命令中使用关键字 f_ID 进行引用输出,其中 ID 是相应fix命令的 ID。需要注意的是,如果你在进行能量最小化的过程中,希望 fix 命令产生的能量和力能够成为优化判据的一部分,那么你必须使用该设置。 - 默认情况下,关键字 temp 指定的 ID 是 fix 命令里默认定义的计算温度的 ID,关键字 press 指定的 ID 是 fix 命令里默认定义的计算压强的 ID,关键字 energy 设置为 no
应用例
xxxxxxxxxxfix_modify 3 temp myTemp press myPressfix_modify 1 energy yes- 关键字
unfixunfix 命令删除之前使用 fix 命令定义的约束
说明
unfix fix-ID
规定进行何种计算:
compute该命令为一组原子执行某种计算。计算出的量是瞬时值,也就是说它们只是原子在当前时间步或迭代步的信息。当然,
compute命令也可以在内部保存体系在之前一个状态的某些信息定义
compute命令的时候并不会执行计算。真正的计算过程是被其他 LAMMPS命令激活的,比如某些fix命令需要计算温度的时候,或这需要产生热力学信息的时候,或者需要dump输出到文件中的时候compute命令可以计算出全局量、单原子量或局部量中某一种类型的量。- 计算产生单原子量的
compute命令在其类型名中含有单词“atom”,比如ke/atom。
- 计算产生局域量的
compute命令在其类型名称中含有单词“local”,比如bond/local。
- 类型名中不包含“atom”或“local”的
compute命令计算产生的是全局量。 - 一个单独的
compute命令只可能会产生这三种类型的量的某一种。
在 LAMMPS 中, compute 命令产生的值有下列的使用方法:
- 计算全局温度或全局压强的 compute 命令产生的结果可以被进行恒温或恒压的
fix命令使用,或者是在创建原子速度的时候被使用。 - 全局量可以使用命令 thermo_style custom or fix ave/time 输出,也可以以 equal 类型或 atom 类型的变量进行引用。
- 单原子量可以使用 dump customor fix ave/spatial 命令进行输出,也可以使用 fix ave/atom 命令对时间进行平均,或使用 compute reduce 命令进行降维,或使用 atom-style 类型的变量进行引用。
- 局域量可以使用 compute reduce 命令进行降维,或者使用 fix ave/histo 命令进行直方图化,或者使用 dump local 命令进行输出。
如果热力学输出需要其他类型的量,可以再添加其他相应的 compute 计算。具体可以参考 thermo_style 命令。
可以计算温度或压强的 fix 命令,比如用于恒温或恒压的 fix 命令,在其内部也可以创建 compute 计算。这些在 fix 命令的页面有进行介绍。
上面列举的所有默认定义的 compute 计算,都可以通过在输入脚本中使用compute 命令定义相应的计算而被替换。这些在 thermo_modify 命令和 fix modify 命令的页面有介绍。
不论是 LAMMPS 内部定义的还是用户自定义的 compute 命令,其设置都可以使用 compute_modify 命令进行修改。
compute 定义的计算可以使用 uncompute 命令删除。
详细说明
compute ID group-ID style argsxxxxxxxxxxID = compute 命令的 IDgroup-ID = 该 compute 命令所作用的原子组的 IDstyle = compute 类型名(最下有列表)args = 特定类型 compute 命令所需要的参数 angle/local – theta and energy of each angle atom/molecule – sum per-atom properties for each molecule body/local – attributes of body sub-particles bond/local – distance and energy of each bond centro/atom – centro-symmetry parameter for each atom cluster/atom – cluster ID for each atom cna/atom – common neighbor analysis (CNA) for each atom com – center-of-mass of group of atoms com/molecule – center-of-mass for each molecule contact/atom – contact count for each spherical particle coord/atom – coordination number for each atom damage/atom – Peridynamic damage for each atom dihedral/local – angle of each dihedral displace/atom – displacement of each atom erotate/asphere – rotational energy of aspherical particles erotate/rigid – rotational energy of rigid bodies erotate/sphere – rotational energy of spherical particles erotate/sphere/atom – rotational energy for each spherical particle event/displace – detect event on atom displacement group/group – energy/force between two groups of atoms gyration – radius of gyration of group of atoms gyration/molecule – radius of gyration for each molecule heat/flux – heat flux through a group of atoms improper/local – angle of each improper inertia/molecule – inertia tensor for each molecule ke – translational kinetic energy ke/atom – kinetic energy for each atom ke/rigid – translational kinetic energy of rigid bodies msd – mean-squared displacement of group of atoms msd/molecule – mean-squared displacement for each molecule pair – values computed by a pair style pair/local – distance/energy/force of each pairwise interaction pe – potential energy pe/atom – potential energy for each atom pressure – total pressure and pressure tensor property/atom – convert atom attributes to per-atom vectors/arrays property/local – convert local attributes to localvectors/arrays property/molecule – convert molecule attributes to localvectors/arrays rdf – radial distribution function g(r) histogram of group of atoms reduce – combine per-atom quantities into a single global value reduce/region – same as compute reduce, within a region slice – extract values from global vector or array stress/atom – stress tensor for each atom temp – 计算一组原子的温度 temp/asphere – temperature of aspherical particles temp/com – temperature after subtracting center-of-mass velocity temp/deform – temperature excluding box deformation velocity temp/partial – temperature excluding one or more dimensions of velocity temp/profile – temperature excluding a binned velocity profile temp/ramp – temperature excluding ramped velocity component temp/region – temperature of a region of atoms temp/sphere – temperature of spherical particles ti – thermodyanmic integration free energy values voronoi/atom – Voronoi volume and neighbors for each atom
应用例
xxxxxxxxxxcompute 1 all tempcompute newtemp flow temp/partial 1 1 0compute 3 all ke/atom- 计算产生单原子量的
compute_modifycompute_modify 命令用来修改过之前定义过的 compute 命令的一个或多个参数 。只有某些特定类型的 compute命令支持修改参数。
详细说明
compute_modify compute-ID keyword value ...xxxxxxxxxxcompute-ID = 需要修改的 compute 命令的 ID可以添加 1 个或多个关键字keyword = extra/dynamic/thermo extra value = N 要减掉的自由度的数量 dynamics value = yes or no 计算温度的时候是否重新计算原子数 thermo value = yes or no 是否考虑 fix 命令计算的势能对总势能的贡献- 关键字
extra用来指定在计算温度时,减掉几个自由度作为规范化因子。 [原文:The extra keyword refers to how many degrees-of-freedom are subtracted (typically from 3N) as a normalizing factor in a temperature computation] 只有那些可以计算温度的 compute 命令可以使用该选项。 The default is 2 or 3 for 2d or 3d systems which is a correction factor for an ensemble of velocities with zero total linear momentum. 如果你需要增加自由度,那么你可以将 extra 设置为负值。命令compute temp/asphere就是这样的一个例子。 - 关键字
dynamic决定在使用 compute 命令计算温度的时候,是否重新计算组内的原子数 N。只有那些可以计算温度的 compute 命令可以使用该选项。默认情况下, N 是一个常量。如果你将一些原子添加到了系统中(比如使用命令fix pourorfix deposit),或者可能存在原子的丢失(比如由于蒸发),那么这个选项可以确保在计算的温度是规范化的。 - 关键字 thermo 决定是否将一些 fix 命令计算的势能加入到该 compute 命令计算出的总势能中。目前只有类型名为
pe的 compute 命令可以使用该选项
- 关键字
uncompute删除之前使用
compute命令定义的计算。它也会删除使用compute_modify命令对该计算所进行的修改uncompute compute-IDvariable
输出选项部分:
thermothermo 命令用来设置在模拟中计算和打印热力学信息(比如温度、能量、压强)的时间步的频率.热力学信息会在时间步为 N 的倍数以及模拟的开始和最后的时候打印
thermo NN:输出热力学信息的频率,可以是变量被打印信息的内容和格式是由命令
thermo_style和thermo_modify控制的。N除了可以是一个数值之外,也可以被指定为 equal 类型的变量,但需要以v_name 的形式引用,其中的 name 就是变量名。在这种情形下,变量会在开始一个 run 之前被计算,从而确定下一个热力学信息输出的时间步。在到了那个时间步时,变量会被再次计算以决定下一个输出的时间步。以此类推。因此变量需要返回时间步的值。函数 stagger(), logfreq(), stride()是与此相关数学变量函数,其他类似的数学函数也可以作为选项被添加。举个例子,下面的命令可以保证在时间步 0,10,20,30,100,200,300,1000,2000 等的时候输出热力学信息
xxxxxxxxxxvariable s equal logfreq(10,3,10)thermo v_sthermo_modify
thermo_style
dump
dump_modify
undump
restartrestart命令在计算的时候,按着特定的模式,以一定的时间步为周期,写二进制的重启动文件。详细说明
xxxxxxxxxxrestart 0restart N filerestart N file1 file2xxxxxxxxxxN:每 N 步写入重启动文件, N 可以是一个变量。file: [单文件模式]重启动文件的文件名。file1/file2: [双文件模式]重启动文件的文件名,交替写重启动文件N 除了可以是一个数值之外,也可以被指定为 equal 样式的变量,但需要以v_name 的形式引用(其中的 name 就是变量名)。在这种情形下,变量会在开始一个 run 之前被计算,从而确定下一个写出重启动文件的时间步。在到了那个时间步时,变量会再次被计算以决定下一个输出的时间步。以此类推。因此变量需要返回的时间步的值。函数 stagger(), logfreq(), stride()是与此相关数学变量函数, 其他类似的数学函数也可以作为选项被添加。
举个例子,下面的命令会从时间步 1100 到 1200 写重启动文件,这对于调试一个在 1163 步出错的模拟来说是非常实用的 :
xxxxxxxxxxvariable s equal stride(1100,1200,1)restart v_s tmp.restart该命令在计算的时候,按着特定的模式(单文件模式和双文件模式),以一定的时间步为周期,写二进制的重启动文件。如果参数是 0,那么就不会写重启动文件。 该命令的两种模式是:
- 单文件模式:如果指定了 1 个文件名,那么程序会创建一系列文件名中包含时间步的重启动文件。
- 双文件模式:如果指定了 2 个文件名,那么程序只会以这两个指定的文件名创建两个重启动文件,并在这两个重启动文件间来回写入重启动信息。
与
dump命令输出文件类似,restart命令在输出重启动文件时,也可以包括两种通配符。- 如果单文件模式的文件名中包括符号“”,那么该符号会被当前的时间步值替换掉。 因此,对于上面例子中的第三个,会创建的重启动文件为: restart.1000.equil, restart.2000.equil, 等。如果单文件模式的文件中不包括“*”,那么时间步值会自动添加在文件名的最后。上面例子中的第二个会创建的重启动文件为:poly.restart.1000, poly.restart.2000等。
- 如果重启动文件的文件名中包括符合“%”,那么每个处理器会写一个文件,并且符号“%”会被处理器的 ID(从 0 到 P-1)替换掉。另外,还会有一个文件,文件名是用“base”代替“%”,其中包含了全局的信息。举例来说,如果使用通配符%,并以 1000时间步的频率写重启动文件,那么会写出的文件有 restart.base.1000,restart.0.1000, restart.1.1000, ……, resta.P-1.1000。使用这种通配符,会创建更小的文件,对于在并行机器中输出和后续的输入都是一种更快的方式。
注意项
注意 1:在一个输入脚本中,你可以两次使用
restart命令,一次是单文件,一次双文件。举例来说,你可以以 100000 步的频率写单文件的重启动文件用来存档,同时你也可以以 1000 步的频率写双文件的重启动文件,将它们作为临时的重启动文件。使用restart 0可以关掉这两种重启动的输出。 注意 2:使用该命令写重启动文件的时间是在时间步是 N 的倍数时,而不是在一个 run 或能量最小化的第一个时间步。如果你希望在一个 run 开始前写重启动文件,那么你可以使用write_restart命令。使用restart命令也不会在一个 run 的最后一个时间步写重启动文件,除非这最后一个时间步恰好是 N 的倍数。但对于能量最小化过程而言,如果其结果收敛的,并且 N 设置>0,那么即便使用 restart 命令,其最后一个时间步也会写入重启动文件。write_restartwrite_restart 命令将模拟的当前状态写入到二进制的重启动文件中
write_restart filefile:要写入重启动信息的文件的文件名在一个耗时很长的模拟中,命令
restart可以按着一定的周期写重启动文件,而命令write_restart可以在完成能量最小化之后,或者按着你的需要随时写出当前状态的重启动文件。与命令
dump类似,该命令的参数 file 中可以包括两个通配符。写出的重启动文件可以使用命令
read_restart读入,从而可以从一个特别的状态重新开始一个模拟。因为重启动文件是二进制的,因此在其他的机器上可能就不可读(二进制文件是依赖于机器的)。在这种情况下,可以使用 tools 目录下提供的restart2data 程序将重启动文件转换为文本式的 data file 格式。命令read_restart和工具restart2data都可以读入由通配符%指定而输出的文件。注意:尽管重启动文件的目的是要从写入重启动文件的位置开始一个模拟,但对一个模拟而言,并不是所有的信息都会存储在重启动文件中。举例来说,原输入脚本中指定的 fix 命令不会存储在重启动文件中,这就要求你在新的输入脚本中需要重新指定你需要用到的 fix 命令。即便有些重启动信息存储在了文件中,就像某些fix 命令,你仍然需要在新的输入脚本中进行定义,从而可以重新使用这些信息。
reset_timestep该命令将时间步的计算器设置为指定值。当使用命令
read_restart读入重启动文件,或在运行一个模拟过程的时候,时间步数就会被设置;但如果你希望重置时间步数为某个数值,可以使用该命令。 命令read_data和create_box会将时间步数设置为 0;read_restart命令会将时间步数设置为重启动文件中写入的值详细说明
xxxxxxxxxxreset_timestep NN:时间步的步数注意项
慎重!
当你定义了一些记录 运行的时间步 ,并基于此进行一些与时间有关的操作时,则不能使用该命令。命令 fix deposit 和 fix dt/reset 就是这样的两个例子。前面一个命令是在指定的时间步里添加原子;后面一个命令是记录累积的时间。 很多 fix 命令会使用当前的时间步来计算相关的量。如果时间步被重新设置的了,这可能会带来难以预测的结果。因为即便时间步被重置了, LAMMPS 也会运行定义的 fix 命令。举例来说,对系统进行恒温控制的命令,比如命令 fix nvt,允许你指定一个目标温度,并通过一定的时间步将温度从从 Tstart 变为 Tstop。但如果你改变时间步数,这可能会带来的结果就是目标温度瞬时被改变。 如果 compute 命令已经计算出了一些量,但在之后使用了重置该命令重置了时间步数,那么它就会清除与 compute 命令相关的标记。这就是说, compute 命令已经计算出来的这些量不能再通过变量进行引用,除非你又重新运行了新的计算。参考变量 variable,了解更多细节。
pair_write该命令将原子对间所定义的势函数,以距离作为自变量,将对应的能量和受力写入到文件中。如果你想绘制势函数曲线或调试势函数,这个命令会比较有用。在写文件的时候,如果文件已经存在了,那么数据会添加到文件的末尾,这样就可以将多个能量-受力 (E-F) 数据块写入到同一文件中。
所写出文件的格式是与 pair_style 命令中 table 类型所需要的输入文件是一样的,其实 keyword 指定的是数据段的名称。写入到文件中的每一行都是由序号(1-N)、原子距离(距离单位)、能量(能量单位)、受力(力单位)构成的。
详细说明:
pair_write itype jtype N style inner outer file keyword Qi Qjitype,jtype2 atom types 指定要计算能量和力的原子类型N# of valuesstyler/rsq/bitmap如果这里设置的 style 是
r,那么会输出 个在 上均匀分布的能量-受力;如果 style 是
rsq,那么会输出 个距离在 上均匀分布的能量-受力。(举个例子,如果 N=7, style=r, inner=1.0, outer=4.0,那么会计算的值有: 。 )
如果 style 是
bitmap[位图],那么会有 个数据按着特定的格式写入到文件 中,其顺序与pair_coeff命令读入pair_styletable 所指定文件的顺序是一致的。 为了能够保证位图数据表的精度,需要设置:,inner小于最近的两个原子 间的距离,outer小于等于势函数的截断距离。inner outerinner and outer cutoff (distance units) 要计算的距离区间filename of file to write values tokeywordsection name in file for this set of tabulated valuesQi,Qj2 atom charges (charge units) (optional)如果要计算势函数的原子对是有电荷的,那么可以为这对原子指定电荷量。如果没有指定,默认会使用 Qi=Qj=1.0
计算中所用到的其他参数由相应的
pair_coeff命令指定
应用例:
xxxxxxxxxxpair_write 1 3 500 r 1.0 10.0 table.txt LJpair_write 1 1 1000 rsq 2.0 8.0 table.txt Yukawa_1_1 -0.5 0.5注意事项:
所有原子对间的力场参数和其他类型的相互作用都必须在使用该命令之前已经被设置好了
受限于程序中原子对间力的计算方式, inner 必须设置为大于 0 的数,即便在 r=0所对应的相互作用是有限值
对于 EAM 势函数而言,该命令只能输出其对势项(pair pot),而不能输出嵌入项(embedbed)
Common Run Command Mannul :
newton:该命令用来开启或关闭对势或键相互作用中的第三运动定律。对于大多数问题而言,将牛顿第三定律设置为开启是一种可以节省 2 倍及以上计算量的做法。具体到是否会更快,则取决于问题的规模、力截断长度、机器的计算/交换比、以及所使用的处理器数量。
详细说明
xxxxxxxxxxnewton flagnewton flag1 flag2x
flag: 开关对势和键相互作用[on/off]flag1:开关对势相互作用[on/off]flag2:开关键相互作用[on/off]将对势相互作用的 flag 设置为 off,那么如果相互作用的两个原子在不同的处理器上,两个处理器都会计算它们的相互作用,所得的关于力的结果信息也不会进行通信。类似的,对于键相互作用,将 flag 设置为 off,那么如果键、角、二面角或不合适的相互作用在 2 个或更多的处理器上,这些相互作用会被每个处理器分别计算。 (注意其速度提升也取决于处理器之间的通讯速度)
所以,对于
run_style respa,如果最内层时间步只计算键相互作用,那么将键相互作用的newton设置为 off 可以避免内层循环的外部通信,从而可能会变得更快。delete_atoms用于删除指定的原子。
详细说明
delete_atoms style args keyword value ...
Chapter 2 Lammps fit command
Overview:
adapt – change a simulation parameter over time addforce – add a force to each atom append/atoms – append atoms to a running simulation aveforce – add an averaged force to each atom ave/atom – compute per-atom time-averaged quantities ave/histo – compute/output time-averaged histograms ave/spatial – compute/output time-averaged per-atom quantities by layer ave/time – compute/output global time-averaged quantities bond/break – break bonds on the fly bond/create – create bonds on the fly bond/swap – Monte Carlo bond swapping box/relax – relax box size during energy minimization deform – change the simulation box size/shape deposit – add new atoms above a surface drag – drag atoms towards a defined coordinate dt/reset – reset the timestep based on velocity, forces efield – impose electric field on system enforce2d – zero out z-dimension velocity and force evaporate – remove atoms from simulation periodically external – callback to an external driver program freeze – freeze atoms in a granular simulation gravity – add gravity to atoms in a granular simulation gcmc – grand canonical insertions/deletions heat – add/subtract momentum-conserving heat indent – impose force due to an indenter langevin – Langevin temperature control lineforce – constrain atoms to move in a line momentum – zero the linear and/or angular momentum of a group of atoms move – move atoms in a prescribed fashion msst – multi-scale shock technique (MSST) integration neb – nudged elastic band (NEB) spring forces nph – constant NPH time integration via Nose/Hoover nph/asphere – NPH for aspherical particles nph/sphere – NPH for spherical particles nphug – constant-stress Hugoniostat integration npt – constant NPT time integration via Nose/Hoover npt/asphere – NPT for aspherical particles npt/sphere – NPT for spherical particles nve – constant NVE time integration nve/asphere – NVE for aspherical particles nve/asphere/noforce – NVE for aspherical particles without forces” nve/body – NVE for body particles nve/limit – NVE with limited step length nve/line – NVE for line segments nve/noforce – NVE without forces (v only) nve/sphere – NVE for spherical particles nve/tri – NVE for triangles nvt – constant NVT time integration via Nose/Hoover nvt/asphere – NVT for aspherical particles nvt/sllod – NVT for NEMD with SLLOD equations nvt/sphere – NVT for spherical particles orient/fcc – add grain boundary migration force planeforce – constrain atoms to move in a plane poems – constrain clusters of atoms to move as coupled rigid bodies pour – pour new atoms into a granular simulation domain press/berendsen – pressure control by Berendsen barostat print – print text and variables during a simulation property/atom – add customized per-atom values reax/bonds – write out ReaxFF bond information recenter – constrain the centerof-mass position of a group of atoms restrain – constrain a bond, angle, dihedral rigid – constrain one or more clusters of atoms to move as a rigid body with NVE integration rigid/nph – constrain one or more clusters of atoms to move as a rigid body with NPH integration rigid/npt – constrain one or more clusters of atoms to move as a rigid body with NPT integration rigid/nve – constrain one or more clusters of atoms to move as a rigid body with alternate NVE integration rigid/nvt – constrain one or more clusters of atoms to move as a rigid body with NVT integration rigid – constrain many small clusters of atoms to move as a rigid body with NVE integration setforce – set the force on each atom shake – SHAKE constraints on bonds and/or angles spring – apply harmonic spring force to group of atoms spring/rg – spring on radius of gyration of group of atoms spring/self – spring from each atom to its origin srd – stochastic rotation dynamics (SRD) store/force – store force on each atom store/state – store attributes for each atom temp/berendsen – temperature control by Berendsen thermostat temp/rescale – temperature control by velocity rescaling thermal/conductivity – 使用 Muller-Plathe 方法计算热导 tmd – guide a group of atoms to a new configuration ttm – two-temperature model for electronic/atomic coupling viscosity – Muller-Plathe 动量交换法计算粘度 viscous – viscous damping for granular simulations wall/colloid – Lennard-Jones wall interacting with finite-size particles wall/gran – frictional wall(s) for granular simulations wall/harmonic – harmonic spring wall wall/lj126 – Lennard-Jones 12-6 wall wall/lj93 – Lennard-Jones 9-3 wall wall/piston – moving reflective piston wall wall/reflect – reflecting wall(s) wall/region – use region surface as wall wall/srd – slip/no-slip wall for SRD particles
Chapter 3 Toolkits Collection : Packmol
Installation:
xxxxxxxxxx./configuremake#生成packmol文件Usage:url
需要提供两类文件 • 输入文件。文件名随意,用来定义各种类型分子如何分布、有多少个。 • 各种要加入的分子的结构文件。一般使用pdb格式,用什么程序产生都可以。
全局设定:
- tolerance [数值]:要求原子间距离不能小于多少,一般设为2.0。Packmol里所有长度的单位都是
- output [文件名]:输出文件的路径
- filetype [类型]:输出文件的格式。默认为pdb,也可以设比如xyz
- seed -1:写这个的话每次运行产生的结构都会不同
- add_box_sides:给输出的pdb中增加CRYST1字段定义盒子,盒子尺寸对应体系中原子最大、最小坐标。如果写诸如add_box_sides 1.2,则会给盒子各方向再增加1.2埃,避免做模拟时有某些边界的原子和镜像盒子里的原子间存在不合理接触
分子设定:
每个下面这种字段设置一种分子如何出现在当前体系中。可以写无数个这种字段。
xxxxxxxxxxstructure [分子结构文件]number [分子数][分子规则]end structure分子规则用来定义分子里的所有原子必须满足的条件,其常用关键词如下:
x
inside cube [xmin ymin zmin d]:要求分子出现在长度为d的立方盒子内,盒子x,y,z最小值为xmin ymin zmininside box [xmin ymin zmin xmax ymax zmax]:要求分子出现在矩形盒子内,盒子两个顶角坐标分别为xmin ymin zmin和xmax ymax zmaxinside sphere [x y z r]:要求分子出现在中心为(x,y,z)半径为r的圆球中。另外,用inside ellipsoid关键词的话还可以要求出现在特定的椭球中inside cylinder [a1 b1 c1 a2 b2 c2 r l]:要求分子出现在圆柱内。圆柱的定义是从(a1,b1,c1)往(a2,b2,c2)方向伸展l长度,半径为r以上inside都可以改为outside,要求不能出现在指定范围内。上面的空间范围要求可以同时使用多个,条件会同时满足。还有一些设置:over plane [a b c d]:要求分子出现位置满足平面方程ax+by+cz-d>=0。比如over plane 1 0 0 10.5就相当于要求出现在x>=10.5埃的区域。over可以改成below来反转条件。constrain_rotation [x/y/z 平均值 最大偏差]:默认情况下分子可以绕x,y,z轴随意旋转任意角度。用这个选项可以设置分子绕x或y或z旋转的情况,可以同时设多个。如constrain_rotation x 20 5代表允许绕着y和z轴随意旋转,而绕着x轴旋转的范围必须在15~25度之间,平均值为20度。fixed [x y z a b c]:用来直接定义分子出现在哪里。此设置代表将分子结构文件里的坐标平移(x,y,z),并绕x,y,z轴分布旋转a,b,c弧度。如果还同时写了center关键词,相当于把分子的中心摆到(x,y,z)位置再旋转
原子设定:
Packmol里还可以要求某种分子的某些特定序号的原子出现的范围必须满足的要求,格式为:
xxxxxxxxxxstructure [分子结构文件][分子规则]atoms [第一批原子序号][原子规则]end atomsatoms [第二批原子序号][原子规则]end atoms...end structure原子规则的设置语法、关键词和分子规则完全一样。
Examples:
构建甲醇盒子:
首先在GaussView或其它可以对分子建模的程序里画一个甲醇,保存为methanol.pdb
用Packmol之前结构不需要非得优化,毕竟GaussView直接画的甲醇的基本几何参数是合理的,而且不管对其做不做优化,等动力学跑起来之后就都一样了。
创建一个名为methanol_box.inp的文本文件,内容如下:
xxxxxxxxxxtolerance 2.0add_box_sides 1.2output methanol_box.pdbstructure methanol.pdbnumber 1000inside cube 0. 0. 0. 50.end structure将methanol.pdb和methanol_box.inp都放到当前目录,运行packmol < methanol_box.inp,经过几秒钟,当前目录下就出现了methanol_box.pdb。

但是1000个甲醇肯定不是Packmol能填充进这个555 nm^3盒子的上限,因为Packmol很容易就产生成功了,而且从图上看还有一些空隙。如果大家希望得到更致密的盒子,可以尝试一点点增加甲醇的number数,看看设到多少的时候Packmol半天也没法成功产生结构或者提示产生失败,Packmol最多能插入多少甲醇你心里就有数了。
构建水+甲醇混合溶液的气液界面体系:
构建一个水+甲醇摩尔比1:1溶液的气液界面体系,界面平行于XY方向,盒子的X、Y长度都为4nm,溶液层厚度是5nm,溶液上下两侧都是真空区,每一侧真空区厚度都是3nm。
water.pdb+methanol.pdb
编辑一个文件mix.inp,内容如下(经soberaeva测试,水和甲醇各插入1000个时盒子已经比较致密了,而且已经需要好多轮循环才能成功产生结构)
xxxxxxxxxxtolerance 2.0add_box_sides 1.2output mix.pdbstructure water.pdbnumber 1000inside box 0. 0. 0. 40. 40. 50.end structurestructure methanol.pdbnumber 1000inside box 0. 0. 0. 40. 40. 50.end structure目前还没有真空区。由于溶液层厚度为5nm,每一侧真空区厚度为3nm,因此盒子Z方向总长度应为5+3+3=11nm。因此我们在VMD里输入pbc set {40 40 110}来设置盒子尺寸为4411 nm^3,这里单位是埃
目前溶剂区域在盒子最底部,我们要将之挪到盒子中间去,也即把所有原子往Z的正方向挪3nm。这可以用VMD的几何变换命令轻易实现。在VMD里输入[atomselect top all] moveby {0 0 30},然后将VMD改成正交视角便于观看(Display - Orthographic),看到下图。

值得一提的是,动力学模拟开始后,肯定最后达到稳定状态时溶液层厚度不可能正好是我们最初期望的5nm,但由于目前已经堆积得比较致密了,应该偏离不太大。如果发现偏离的程度超过自己能接受的程度,可以在建模时再适当增加点分子数、在packmol建模时让盒子在Z方向稍微大一点。
氯仿+甲醇+富勒烯体系:
构建一个半径1.5nm的溶剂球,甲醇和氯仿各一个半球区域,并且富勒烯正好出现在球中央。
chloroform.pdb和C60.pdb都是GaussView直接构建的,后者在GaussView的片段库里直接就有。
x
tolerance 2.0#由于当前模拟的是孤立体系,所以没有用add_box_sides关键词来产生盒子信息。output mix.pdb#对于氯仿和甲醇,inside sphere要求它们只能出现于距离(2,2,2)半径1.5 nm的区域内,由于二者的inside box设定范围正好相接,因此这两种分子会分布出现在Z=2nm平面的上、下两侧。structure chloroform.pdbnumber 200inside box 0. 0. 0. 40. 40. 20.inside sphere 20. 20. 20. 15.end structurestructure methanol.pdbnumber 200inside box 0. 0. 20. 40. 40. 40.inside sphere 20. 20. 20. 15.end structure#我们假定球的中心在(2,2,2) nm的位置,因此直接用center和fixed关键词将C60的中心定位在这个地方。structure C60.pdbnumber 1centerfixed 20. 20. 20. 0. 0. 0.end structure
构建水球+十二烷基磺酸钠:
这个例子主要用来体现怎么使用原子设定。十二烷基磺酸钠(SDS)的头部基团是亲水的,烷烃尾巴是疏水的。此例我们要构建一个半径为20埃的水球,让100个SDS绕着它分布,SDS的头部基团都埋在水球里,而且让烷烃尾巴的方向基本上垂直于水球的界面,最终构建出的结构应该类似于海胆,SDS就是海胆的刺。
我们先用GaussView画一个SDS。由于钠离子的位置放在哪可能凭直觉不好确定,因此画完之后可以做一下几何优化,比如用Gaussian在PM7半经验方法下粗略优化后得到如下结构,看起来很合理

xxxxxxxxxxtolerance 2.0output mix.pdbstructure water.pdbnumber 1200inside sphere 0. 0. 0. 20.end structure#安置SDS的这一段值得提一下。我们让42号原子,即钠离子必须出现在距离水球中心15埃范围以内structure SDS.pdbnumber 100atoms 42inside sphere 0. 0. 0. 15.end atoms#而让1号原子,即烃链最末端的碳原子必须距离水球中心30埃以上。atoms 1outside sphere 0. 0. 0. 30.end atomsend structure#由于在SDS中这俩原子距离为18埃,仅略大于30-15=15埃,因此这么定义可以起到让SDS链垂直于水球界面的效果。
Note:
- Packmol的执行过程实际上是个循环过程,如果你当前的空间范围设定比较简单,Packmol很快就会顺利产生最终结构。空间约束设定得越多、越复杂,往往成功产生结构需要的循环次数越多,总耗时越高。如果你把空间范围约束得过度复杂甚至相互矛盾,或者要加入的分子数较多但允许分子出现的空间范围相对而言过度狭小,那么Packmol会反复循环,一直也收敛不了,或者最终宣告产生失败。碰到这种情况,应当将约束条件适当放宽、简化,或者设的分子数少一点再试。
- 一般情况下,用Packmol构建的体系肯定是比较松散的,或者说其密度肯定显著低于真实密度,因为此时还没有考虑分子间相互作用、构象的变化从而使得分子间能够接触得更紧密。但这没关系,在NPT下跑一下MD就好了,盒子尺寸会自发进行调节。而直接用Packmol产生的结构做NVT模拟一般是无意义的,因为跑起来之后分子会自发紧密聚集,其它地方就成了真空区了,和实际不符。
- 用Packmol实现构建含有Na+、Cl-离子的溶液体系是可以的。但是肯定没有用GROMACS里专用的gmx genion命令好,因为gmx genion在插入时会考虑体系电荷分布,会把阳离子和阴离子分别加入到静电势较负和较正的地方,这样比较合理。而Packmol则没有考虑这点,并且有可能比如产生的结构里恰好Na+出现在显著带正电荷的区域,这可能会令模拟初期稳定性较差(此时需要用较谨慎的模拟参数,比如小步长)。
- 虽然也可以用Packmol构建蛋白质、核酸浸在溶剂环境中的体系,但是这样做明显不如用动力学程序自带的专用做这种事情的程序好,因为Packmol产生的水的密度偏低,水的分布特征和实际体相水相差较大,可能NPT模拟起来之后盒子变形、收缩得厉害,出现溶质与其镜像最近距离太近之类问题。而如果用比如GROMACS里的gmx solvate命令给蛋白质/核酸加溶剂,由于是直接用事先已经做NPT平衡好的溶剂盒子(比如加水一般用自带的spc216.gro)通过平移复制来填充真空区,加的溶剂的分布状态明显理想得多得多。
- GROMACS有一个命令叫gmx insert-molecules,可以由用户提供分子结构文件、指定插入盒子的分子数,然后试图将这些分子都插入盒子(但如果设的数目太多,会能插入多少就插入多少)。此命令远没有Packmol那么灵活,它能做的事用Packmol都可以实现,而且Packmol会通过优化算法试图让分子尽可能好地堆叠,因此对于同样的盒子范围,靠Packmol最多能插入的分子数比用gmx insert-molecules明显要多,故能得到更紧凑的结构。
- Packmol原理上也可以产生磷脂双分子膜,乃至膜蛋白这样的体系的,但是这需要设置很多约束条件才能产生靠谱的结果,而且耗时很高,结果也不理想。比如磷脂之间或者磷脂与蛋白之间会有好多空隙,这会导致模拟刚开始就有水大量跑进疏水区,后续模拟将毫无意义。构建磷脂膜最推荐用sobereva写的genmixmem程序,速度很快,用着很方便,结合GROMACS模拟磷脂体系特别容易,详见《生成混合组分的磷脂双层膜结构文件的工具genmixmem》(http://sobereva.com/245)。
- 如果你要建模的是分子团簇,用于构型搜索之类目的,用sobereva开发的genmer比使用Packmol好得多,一次可以产生一大批,输入文件写起来省事得多,参见《genmer:生成团簇初始构型结合molclus做团簇结构搜索的超便捷工具》(http://bbs.keinsci.com/thread-2369-1-1.html)。
- 如果你用Packmol建模的目的是之后用GROMACS做模拟,别忘了GROMACS的拓扑文件里[molecules]字段里的分子按顺序完全展开后,原子的顺序必须和结构文件完全相同。为保证这一点,用于Packmol建模的分子结构文件里的原子顺序首先就得和这个分子的拓扑信息(具体来说是[atoms]字段)相对应。
Reference: http://sobereva.com/473
Chapter 3 Toolkits Collection : atomsk
Atomsk is a free, Open Source command-line program dedicated to the creation, manipulation, and conversion of data files for atomic-scale simulations. It supports the file formats used by many programs, including:
- Visualization and analysis softwares: Atomeye, OVITO, VESTA, XCrySDen;
- Ab initio calculation softwares: Quantum Espresso, VASP, SIESTA;
- Classical force-field simulation softwares: DL_POLY, GULP, IMD, LAMMPS, XMD;
- TEM image simulation softwares: Dr Probe, JEMS, QSTEM.
Atomsk proposes options for applying elementary transformations: duplicate, rotate, deform, insert dislocations, merge several systems, create bicrystals and polycrystals, etc. These elementary tools can be combined to construct and shape a wide variety of atomic systems. To learn more, you are invited to read the documentation, and to follow the tutorials.
If you use Atomsk in your work, the citation of the following article will be greatly appreciated:
- "Atomsk: A tool for manipulating and converting atomic data files" Pierre Hirel, Comput. Phys. Comm. 197 (2015) 212-219 doi:10.1016/j.cpc.2015.07.012
Basics:
Install and run Atomsk:
win: unzip + add path
Linux: lscpu + add path
convert file:
atomsk Al_unitcell.xsf cfg lammps vaspOptions:
atomsk Al_unitcell.xsf -duplicate 2 2 3 Al_supercell.cfgCreation:
atomsk --create fcc 4.046 Al aluminium.xsfatomsk --create rocksalt 5.64 Na Cl xsfatomsk --create hcp 3.21 5.213 Mg Mg.xsfatomsk --create hcp 3.21 5.213 Mg -orthogonal-cell Mg.xsfatomsk --create graphite 3.21 5.213 C graphite.xsfatomsk --create nanotube 2.6 8 0 C CNT.cfgatomsk --create nanotube 2.6 8 0 C -shift 0.5*box 0.5*box 0.0 CNT.cfgatomsk --merge 2 CNT2.cfg CNT.cfg MWNT.cfgatomsk --merge 2 CNT2.cfg CNT.cfg MWNT.cfg -shift 0.5*box 0.5*box 0.0atomsk --create nanotube 2.6 8 0 B N BNNT.cfgShift:
atomsk aluminium.xsf -shift 10 10 10 Al_shift.xsfIonic charge:
atomsk --create rocksalt 5.64 Na Cl -properties charge.txt cfgxxxxxxxxxx# Charges for NaCl crystalchargeNa 1.0Cl -1.0Create a cubic lattice with a specific crystal orientation:
atomsk --create fcc 3.597 Cu orient [110] [-110] [001] Cu.cfgatomsk --create hcp 3.21 5.213 Mg orient [0-110] [0001] [2-1-10] xsfCrystal rotation:
atomsk initial.xsf -rotate Z 33 final.xsfatomsk Fe.xsf -orient [100] [010] [001] [121] [-101] [1-11] Fe_orient.cfgVisualize:
atomsk 9002806.cif vestaTransform to lammps:
atomsk 9002807.cif lammpsatomsk 9002806.cif -orient [110] [-110] [001] [100] [010] [001] SrTiO3_rotated.cfgatomsk NaCl.xsf -add-shells all NaCl_shells.xsfatomsk MD0.xyz MD1.xyz MD2.xyz MD3.xyz MD4.xyz MD5.xyz lammps
Construct geometric shapes:
Remove atoms:
atomsk NaCl_supercell.xsf -remove-atoms Cl final.cfgatomsk NaCl_supercell.xsf -remove-atoms 417 final.cfgatomsk NaCl_supercell.xsf -remove-atoms 417 -remove-atoms 210 final.cfg(from 417 to 210, if remove small No. atom first, all the list No will change!)atomsk NaCl_supercell.xsf -select Cl -remove-atoms select final.cfgSelect atoms at random:
atomsk NaCl_supercell.xsf -select random 30 Na -remove-atoms select final.cfgatomsk NaCl_supercell.xsf -select random 1.5% Na -remove-atoms select final.cfgatomsk NaCl_supercell.xsf -select random 30 any -remove-atoms select final.cfgCut plane:
atomsk Si_supercell.cfg -cut above 30 x Si_cut.cfgatomsk Si_supercell.cfg -cut above 40 [111] Si_cut.cfguse after select:
atomsk Si_supercell.cfg -select above box-5 y -cut above 30 x Si_cut.cfgCreate a cell with the desired orientation:
atomsk --create fcc 4.08 Au -duplicate 40 40 40 Au_cell.xsfNanopillar of cubic shape:
xxxxxxxxxxatomsk Au_cell.xsf \-select out box 0.2*box -INF 0.2*box 0.8*box INF 0.8*box \-remove-atom select \nanopillar.cfgNanopillar of cylindrical shape:
xxxxxxxxxxatomsk Au_cell.xsf \-select out cylinder Y 0.5*box 0.5*box 40 \-cut above 20 Y \nanopillar.cfgArray of nanopillars:
atomsk nanopillar.cfg -duplicate 3 1 3 array.cfgSphere of Silicon:
The supercell of silicon:
atomsk --create diamond 5.431 Si -duplicate 30 30 30 Si_supercell.xsfCarving a sphere:
xxxxxxxxxxatomsk Si_supercell.xsf -select out sphere 0.5*box 0.5*box 0.5*box 70 \-rmatom select Si_sphere.cfgCreate a half-sphere:
atomsk Si_sphere.cfg -cut below 0.5*box Y Si_halfsphere.cfgFastest way:
xxxxxxxxxxatomsk --create diamond 5.431 Si \-duplicate 30 30 30 \-select out sphere 0.5*box 0.5*box 0.5*box 70 \-rmatom select \-cut below 0.5*box Y \Si_final.cfg
Half-Sphere of Cu on Ta Substrate:
The tantalum (Ta) substrate:
atomsk --create bcc 3.31 Ta orient [1-10] [001] [110] \ -duplicate 40 60 10 bccTa_substrate.cfgThe copper (Cu) half-sphere:
xxxxxxxxxxatomsk --create fcc 3.61 Cu orient [11-2] [1-10] [111] \-duplicate 40 60 20 \-select out sphere 0.5*box 0.5*box 0.0 60 \-rmatom select fccCu_halfsphere.cfgMerge the two systems:
atomsk --merge Z 2 bccTa_substrate.cfg fccCu_halfsphere.cfg CuTa_final.cfg
Combine selections:
The option "-select":
xxxxxxxxxxatomsk Al.cfg -select in sphere 0.35*box 0.5*box 0.5*box 50 \-select invert -rmatom select groupA.cfgxxxxxxxxxxatomsk Al.cfg -select in sphere 0.65*box 0.5*box 0.5*box 50 \-select invert -rmatom select groupB.cfgxxxxxxxxxxatomsk Al.cfg -select in sphere 0.35*box 0.5*box 0.5*box 50 \-select in sphere 0.65*box 0.5*box 0.5*box 50 \-select invert -rmatom select test.cfgAdd atoms to the selection : (A) OR (B)
xxxxxxxxxxatomsk Al.cfg -select in sphere 0.35*box 0.5*box 0.5*box 50 \-select add in sphere 0.65*box 0.5*box 0.5*box 50 \-select invert -rmatom select add.cfgRemove atoms from the selection : (A) AND NOT(B):
xxxxxxxxxxatomsk Al.cfg -select in sphere 0.35*box 0.5*box 0.5*box 50 \-select rm in sphere 0.65*box 0.5*box 0.5*box 50 \-select invert -rmatom select rm.cfgKeep only atoms that belong to both groups : (A) AND (B)
xxxxxxxxxxatomsk Al.cfg -select in sphere 0.35*box 0.5*box 0.5*box 50 \-select intersect in sphere 0.65*box 0.5*box 0.5*box 50 \-select invert -rmatom select intersect.cfgKeep atoms that belong to either group, but not both : (A) XOR (B)
xxxxxxxxxxatomsk Al.cfg -select in sphere 0.35*box 0.5*box 0.5*box 50 \-select xor in sphere 0.65*box 0.5*box 0.5*box 50 \-select invert -rmatom select xor.cfg
Select Atoms According to a Grid:
atomsk --create fcc 4.046 Al -duplicate 40 40 3 Al_supercell.xsfWrite a mask in a text file:
xxxxxxxxxx000000000000000000000011000001111000001111000011001100000011000000011000000011000000110000000111100011111100000000000000000000000000000000000000Remove or substitute atoms according to the mask:
atomsk Al_supercell.xsf -select grid mask.txt -rmatom select Al_12.cfgatomsk Al_supercell.xsf -select grid mask.txt -cut above 4.1 Z Al_12.cfgatomsk Al_supercell.xsf -select grid grid_1.txt -substitute Al Au Al_12.cfgConstruct 3-D grids:
xxxxxxxxxx5 5 41000001000001000001000001000000010001110001000000000100001001111100100001000000100010001000100010000
Select Atoms According to a 3-D Shape:
Design or download a STL file:
- you may design one yourself, using 3-D softwares like Tinkercad, Blender, Google Sketchup, or similar softwares;
- or, you may download an existing 3-D model from an online Website, like Cults3D, Pinshape, Thingiverse, or other sources.
atomsk --create fcc 4.046 Al -duplicate 40 40 40 Al_supercell.xsfatomsk Al_supercell.xsf -select stl center Dog_lowpoly_flowalistik.STL -select invert -rmatom select Dog.cfg
Point Defects:
atomsk --create rocksalt 5.64 Na Cl -duplicate 10 10 10 NaCl_supercell.xsfVacancies:
atomsk NaCl_supercell.xsf -select random 30 Na -remove-atoms select final.cfg``atomsk NaCl_supercell.xsf -select random 1% Na -remove-atoms select final.cfg``atomsk NaCl_supercell.xsf -select 417 -remove-atom select final.cfgSubstitute atoms:
atomsk NaCl_supercell.xsf -substitute Na Ag final.cfgatomsk NaCl_supercell.xsf -select 417 -substitute Na Ag final.cfgatomsk NaCl_supercell.xsf -select random 30 Na -substitute Na Ag final.cfgatomsk NaCl_supercell.xsf -select random 1% Na -substitute Na Ag final.cfgInsert new atoms:
atomsk NaCl_supercell.xsf -add-atom Si at 23 34 45 final.cfgatomsk NaCl_supercell.xsf -add-atom Si near 417 final.cfgatomsk NaCl_supercell.xsf -add-atom Si random 21 final.cfg
Stacking Fault:
- Crystallographic orientation of the system:
atomsk --create fcc 4.046 Al orient [-110] [111] [11-2] -duplicate 1 8 1 Al_cell.xsf - Shift the top crystal:
atomsk Al_cell.xsf -shift above 0.5*box Y 1.0 0.0 0.0 Al_SF.xsf - Alternative way with option "-select":
atomsk Al_cell.xsf -select above 0.5*box Y -shift 1.0 0.0 0.0 Al_SF.xsf - Atoms out of the box:
atomsk Al_cell.xsf -shift above 0.5*box Y 1.0 0.0 0.0 -wrap Al_SF.xsf
- Crystallographic orientation of the system:
Twin Boundary:
- Crystallographic orientation of the system:
atomsk --create fcc 4.02 Al orient [11-2] [111] [-110] -duplicate 1 8 1 Al_cell.xsf - Apply mirror symmetry:
atomsk Al_cell.xsf -mirror 0 Y -wrap Al_mirror.xsf - Stack the two crystals:
atomsk --merge Y 2 Al_cell.xsf Al_mirror.xsf Al_final.cfg
- Crystallographic orientation of the system:
Screw Dislocation in Aluminium:
Crystallographic orientation of the system:
atomsk --create fcc 4.046 Al orient [1-12] [-111] [110] -duplicate 40 20 1 Al_supercell.xsfIntroduce a screw dislocation:
atomsk Al_supercell.xsf -dislocation 0.51*box 0.501*box screw Z Y 2.860954 Al_screw.xsf cfgDeal with boundary conditions:
xxxxxxxxxx# Fcc Al oriented X=[1-12] Y=[-111] Z=[110].CRYSTALPRIMVEC198.21270999 0.00000000 1.430477000.00000000 140.15755135 0.000000000.00000000 0.00000000 2.86095404CONVVEC198.21270999 0.00000000 1.430477000.00000000 140.15755135 0.000000000.00000000 0.00000000 2.86095404PRIMCOORD(...)Construct a quadrupole of dislocations:
xxxxxxxxxxatomsk --create fcc 4.046 Al orient [1-12] [-111] [110] \-duplicate 40 30 1 \-dislocation 0.251*box 0.251*box screw Z Y 2.860954 \-dislocation 0.751*box 0.251*box screw Z Y -2.860954 \-dislocation 0.251*box 0.751*box screw Z Y -2.860954 \-dislocation 0.751*box 0.751*box screw Z Y 2.860954 \Al_quadrupole.cfg
Edge Dislocation in Aluminium:
Crystallographic orientation of the system:
atomsk --create fcc 4.046 Al orient [110] [-111] [1-12] Al_unitcell.xsf``atomsk Al_unitcell.xsf -duplicate 60 20 1 Al_supercell.xsfIntroduce an edge dislocation by inserting a half-plane:
atomsk Al_supercell.xsf -dislocation 0.51*box 0.501*box edge Z Y 2.860954 0.33 Al_edge.cfgIntroduce an edge dislocation without changing the total number of atoms:
atomsk Al_supercell.xsf -dislocation 0.51*box 0.501*box edge2 Z Y 2.860954 0.33 Al_edge2.cfgCreate a dislocation by superimposing two crystals:
atomsk Al_unitcell.xsf -duplicate 40 10 1 -deform X 0.0125 0.0 bottom.xsfatomsk Al_unitcell.xsf -duplicate 41 10 1 -deform X -0.012195122 0.0 top.xsfatomsk --merge Y 2 bottom.xsf top.xsf Al_edge3.xsf cfg
Dislocations in Anisotropic Medium:
Crystallographic orientation of the system:
atomsk --create bcc 2.856 Fe orient [121] [-101] [1-11] xsfGenerate the system containing a screw dislocation:
xxxxxxxxxxatomsk --create bcc 2.856 Fe orient [121] [-101] [1-11] \-duplicate 20 30 1 \-prop elastic.txt \-disloc 0.501*box 0.501*box screw z y 2.47336855321 \Fe_dislo.cfgxxxxxxxxxxatomsk --create bcc 2.856 Fe orient [1-11] [-101] [121] \-duplicate 20 30 1 \-prop elastic.txt \-disloc 0.501*box 0.501*box edge z y 2.47336855321 0.0 \Fe_edge.cfg
Dislocation and Inclusion:
The aluminium (Al) matrix with the screw dislocation:
xxxxxxxxxxatomsk --create fcc 4.046 Al orient [1-12] [-111] [110] \-duplicate 50 30 30 \-dislocation 0.251*box 0.501*box screw Z Y 2.860954 \-select in sphere 0.75*box 0.5*box 0.5*box 20 \-remove-atoms select \fccAl_matrix.cfgThe silicon (Si) inclusion:
xxxxxxxxxxatomsk --create diamond 5.431 Si \-duplicate 60 40 40 \-select out sphere 185.824 105.118 42.914 20 \-remove-atoms select \Si_sphere.cfgxxxxxxxxxxatomsk --create diamond 5.431 Si \-duplicate 10 10 10 \-shift -0.5*box -0.5*box -0.5*box \-select out sphere 0 0 0 20 \-remove-atoms select \-select none \-shift 185.824 105.118 42.914 \Si_sphere.cfg xsf
Merge the two systems:
atomsk --merge 2 fccAl_matrix.cfg Si_sphere.cfg AlSi_final.cfg
Polycrystals:
atomsk --create fcc 4.046 Al aluminium.xsfBasics of Voronoi tessellation
Atomsk uses a Voronoi tesselation to construct polycrystals, as illustrated in the figure below.
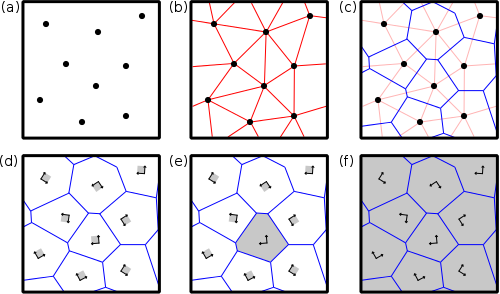
(a) Nodes (black dots) are introduced at given positions inside the simulation box.
(b) Nodes are linked with their neighbors (red lines). Periodic boundary conditions are used.
(c) The normal to the red lines are found (blue lines). These blue lines define the contours of the future grains, i.e. the grain boundaries.
(d) Atomic "seeds" (for instance unit cells) are placed at the positions of the nodes, with the given crystal orientation.
(e) A seed is expanded in the three directions of space. Atoms that are outside of the grain are removed.
(f) After all seeds have been expanded and cut inside their respective grains, one obtains the final polycrystal.
This illustrates the generation of Voronoi polycrystal in 2-D, but can be generalized to the 3-D case.
Polycrystal with grains at random positions and orientations:
atomsk --polycrystal aluminium.xsf polycrystal.txt final.cfgBoundary conditions:
atomsk --polycrystal aluminium.xsf polycrystal.txt bcc.cfgControl the position and/or orientation of each grain:
atomsk --polycrystal aluminium.xsf polycrystal.txt final.xsf cfg
Multi-Phase Polycrystals:
The copper polycrystal:
atomsk --create fcc 3.61 Cu Cu_unitcell.xsfatomsk --polycrystal Cu_unitcell.xsf polycrystal.txt Cu_polycrystal.cfgatomsk --polycrystal Cu_unitcell.xsf polycrystal.txt Cu_polycrystal.cfg \ -select prop grainID 1 -rmatom select -select prop grainID 6 -rmatom selectThe tungsten polycrystal:
atomsk --create bcc 3.16 W W_unitcell.xsfxxxxxxxxxxatomsk --polycrystal W_unitcell.xsf polycrystal.txt W_polycrystal.cfg \-select prop grainID 2:5 -rmatom selectMerging the two previous systems:
atomsk --merge 2 Cu_polycrystal.cfg W_polycrystal.cfg final_polycrystal.cfg
Grain Boundaries:
Basics: the example of the symmetric tilt grain boundary
Tilt grain boundaries are a specific type of grain boundary, where the two grains are rotated around the same crystallographic axis. When the two grains are rotated by opposite angles, the tilt grain boundary is symmetric.
The steps to construct a symmetric tilt grain boundary are as follow (here we consider the Cartesian Y axis as being normal to the grain boundary, and Z normal to the figure):
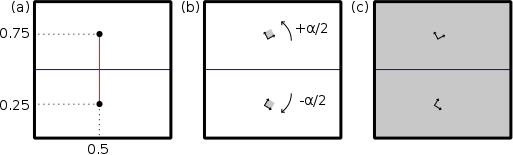
(a) Insert two nodes at the reduced coordinates (0.5,0.25,0) and (0.5,0.75,0). By doing so, the grain boundary is located at the center of the final cell (horizontal blue line). Note that, because of periodic boundary conditions, there will also be a grain boundary at the bottom of the box, and one at the top.
(b) Insert the seeds at the positions of the nodes. Rotate one seed by an angle +α/2 and the other one by -α/2 around Z, so that the desorientation between the two grains is α.
(c) Duplicate the seed in all directions of space, and cut each grain at the grain boundary.
Constructing a grain boundary in aluminium:
atomsk --create fcc 4.046 Al aluminium.xsfatomsk --polycrystal aluminium.xsf polyX.txt polycrystal.cfg
Chapter 4 Toolkits Collection: Potfit
A bridge connecting DFT and MD
- Installation:
- Usages: